This article is a note for:
[https://doi.org/10.1051/parasite/2018029]
| Issue |
Parasite
Volume 25, 2018
|
|
|---|---|---|
| Article Number | 30 | |
| Number of page(s) | 17 | |
| DOI | https://doi.org/10.1051/parasite/2018028 | |
| Published online | 28 May 2018 | |
Research Article
Analysis of Dipylidium caninum tapeworms from dogs and cats, or their respective fleas
Part 1. Molecular characterization of Dipylidium caninum: genetic analysis supporting two distinct species adapted to dogs and cats
Analyse des ténias Dipylidium caninum des chiens et des chats, ou de leurs puces respectives
Partie 1. Caractérisation moléculaire de Dipylidium caninum : analyse génétique soutenant deux espèces distinctes adaptées aux chiens et aux chats
1
Boehringer Ingelheim Animal Health,
29 Av Tony Garnier,
69007
Lyon, France
2
Clinomics, P.O. Box 11186, Universitas,
9321,
Bloemfontein, South Africa
3
Boehringer Ingelheim Animal Health, Kathrinenhof Research Centre,
Walchenseestr. 8-12,
83101
Rohrdorf, Germany
4
École Nationale Vétérinaire de Maisons-Alfort,
94704
Maisons-Alfort Cedex, France
5
Clinvet, P.O. Box 11186, Universitas,
9321,
Bloemfontein, South Africa
* Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
25
January
2018
Accepted:
21
April
2018
Abstract
A 28S rDNA PCR detection assay was previously developed to identify Dipylidium caninum DNA inside single fleas collected from both cats and dogs. Sequence analysis of the 28S rDNA fragment indicated two genetically distinct variations of the target region. The two genotypes, so-called “D. caninum canine genotype” and “D. caninum feline genotype”, based on host origin, are further investigated and described in this paper. Restriction fragment length polymorphism (RFLP) analysis and hydrolysis probe-based genotyping assays were developed and validated for genotyping D. caninum DNA. The complete mitochondrial (mt) genome of the “feline genotype” was sequenced and compared to the D. caninum mt genome available in GenBank. The molecular characterization of D. caninum isolates collected from infected fleas, and also proglottids collected from dogs and cats, confirmed the existence of two distinct genotypes. These genotypes are related to host origin (dogs or cats), irrespective of their geographical origin, and they present a biological adaptation to their respective host, as confirmed by the comparison of biological development and host preference in another study. The genetic differences (Part 1, present paper) and biological observations (Part 2, in this journal) enabled us to suggest the existence of two distinct species within D. caninum, which will have to be clarified.
Résumé
Un test PCR de détection de fragment de l’ADN ribosomal de Dipylidium caninum dans des puces récoltées sur chiens et chats a été précédemment développé. L’analyse des séquences de l’ADNr 28S a démontré la présence de deux génotypes distincts. Ces deux génotypes, appelés génotype canin et génotype félin sur la base de l’origine des isolats, sont étudiés et présentés dans cet article. L’hydrolyse de fragments d’ADN et l’analyse du polymorphisme de taille des sites de restriction ont été mise au point et validées pour génotyper l’ADN de D. caninum. Le séquençage complet du génome mitochondrial du génotype félin a été réalisé et comparé au génotype canin, dont le génome mitochondrial est disponible dans GenBank. L’analyse moléculaire des isolats de D. caninum collectés à partir de puces infectées, ou de proglottis issus de chiens et chats infestés confirme l’existence de deux génotypes distincts. Ces génotypes sont liés à l’hôte d’origine, chien ou chat, quelle que soit leur origine géographique, et ils présentent une adaptation biologique à leur hôte d’origine, comme confirmé dans une autre étude. Les différences génétiques (Partie 1, cet article) et les observations biologiques (Partie 2, dans ce journal) permettent de suggérer l’existence de deux espèces au sein de D. caninum, ce qui devra être clarifié.
Key words: Dipylidium caninum / mitochondrial genome / Ctenocephalides felis / dogs / cats / genotypes
Michel Labuschagne and Frédéric Beugnet are first authors at equal position and contributed equally to this manuscript.
© M. Labuschagne et al., published by EDP Sciences, 2018
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Dipylidium caninum (Linnaeus, 1758), a globally distributed cestode, infects domestic cats and dogs [8], wild canids and felids [7,10], and occasionally humans [15]. In 1758, Linnaeus recognized the parasite and named it Taenia canina. In 1863, Leuckart erected the genus Dipylidium, but it was not until 1893 that the internal anatomy and observations of the life history of Dipylidium caninum was described by Diamare (in [16]). The cat flea, Ctenocephalides felis, is considered the main intermediate host of D. caninum [5]. Furthermore, C. felis has the ability to infest both dogs and cats. The dog flea (Ctenocephalides canis) also acts as an intermediate host, but exceptionally infests cats. The life cycle of D. caninum can be summarized as follows: flea larvae ingest eggs of D. caninum, followed by development of the egg to the metacestode stage inside the flea. When a canine or feline host ingests adult fleas infected with suitably developed metacestodes, the parasite establishes in the small intestine of its definitive host.
Several species belonging to the genus Dipylidium have historically been suggested based on morphological observations [16]. However, significant overlap in morphological traits led to recognition of a single species, namely D. caninum [16,18]. As a result, the genus Dypilidium is currently considered monotypic. However, in the absence of distinguishing morphological characters or significant overlap thereof, modern molecular techniques often allow us to differentiate hidden genetic lineages and cryptic species [e.g. 1,6,12,17,19]. A molecular approach would potentially be highly beneficial in investigating and confirming species status within the genus Dipylidium.
In 2014, Beugnet et al. [3] investigated the prevalence of D. caninum in fleas from client-owned cats and dogs in Europe, using a new PCR detection assay targeting a region within the 28S rDNA. The results confirmed the spread of D. caninum sensu lato in fleas of dogs and cats throughout Europe. Preliminary analyses indicated genetic differences between D. caninum metacestodes in fleas collected from dogs and cats, respectively. These preliminary analyses are described in the present article as well as all further molecular assessments that were performed in order to confirm the existence of multiple D. caninum genotypes.
Original sequence analysis of PCR products from Dipylidium infected fleas collected in 2011 and 2012 (Beugnet et al., 2014 [3]) indicated two 28S rDNA sequence variants of the target region. This preliminary analysis suggested that a host-specific preference may be applicable, and hence the two distinct 28S rDNA sequence variants were defined as the so-called “D. caninum canine genotype” and the “D. caninum feline genotype” [previously unpublished, see Tables 1–Tables 1 to 3]. The D. caninum canine genotype was found in > 95% cases in infected C. felis fleas collected on dogs and 100% of C. canis infected fleas, whereas the D. caninum feline genotype was identified in > 95% of C. felis infected fleas collected on cats. It was thus decided to further investigate these differences, with specific reference to possible genotype-host associations.
The present paper reports the several steps of analysis since the original finding. Firstly, the development of restriction fragment length polymorphism (RFLP) analysis and hydrolysis probe-based genotyping assays, for genotyping D. caninum DNA. Secondly, a sensitive, non-invasive nucleic acid detection assay applied for the detection of D. caninum DNA in faeces. And finally, the complete sequencing of the mt genome from a feline host to compare the “D. caninum feline genotype” and the D. caninum mt genome available in GenBank.
The objectives of this study were thus to develop the necessary assays for genotyping D. caninum DNA to discriminate between the two D. caninum genotypes. These comparisons allowed the identification and breeding of the two genotypes, in order to further evaluate their biological differences (see Beugnet et al., 2018, part 2, [4]).
Details on two distinct 28S rDNA sequence variants (“canine” and “feline”) defined from fleas infected by Dipylidium caninum, obtained from PCR products collected by Beugnet et al. [3] from Europe and South Africa.
Details on two distinct 28S rDNA sequence variants (“canine” and “feline”) defined from fleas infected by Dipylidium caninum, received from the United States of America.
Details on two distinct 28S rDNA sequence variants (“canine” and “feline”) defined from non-invasive anal swabs collected from cats and dogs infected by Dipylidium caninum in South Africa.
Materials and Methods
Parasites and DNA extraction
28S rDNA PCR detection of D. caninum was performed on 6116 crude flea (C. felis, C. canis and Pulex irritans) extracts as described by Beugnet et al [3]. A total of 192 D. caninum-positive DNA extracts were included in the present genotype analysis. A total of 57 D. caninum-positive samples were subjected to DNA sequencing. Subsequently, all positive samples (Table 1) were subjected to RFLP (restriction fragment length polymorphism) analysis.
In addition, a total of 55 fleas collected from cats were obtained from Mike Lappin (Colorado State University, USA) and 9 of the PCR positive samples were subjected to sequencing (Table 2). Adult fleas (n = 100) were also obtained from New Zealand and 2 of the PCR positive samples were subjected to sequencing. All adult fleas were supplied in 70% (v/v) ethanol.
Five adult D. caninum tapeworms were also obtained from various sources, including 2 worms from Clinvet International (South Africa), 1 worm from École Nationale Vétérinaire de Maisons-Alfort (France), and 2 worms from Guangxi University Animal Hospital (China). All adult worms were supplied in 70% (v/v) ethanol. Genomic DNA was isolated from the adult worm proglottids using the GeneJet Genomic DNA isolation kit (Thermo Fisher Scientific), according to the manufacturer’s recommendation for tissue samples.
The original D. caninum strain maintained at Clinvet International research centre on cats and fleas was included. A new Dipylidium sp. strain originating from proglottids collected on dogs in the village near the research centre was also included, genotyped, and maintained on dogs. The two identified genotypes are thus maintained on dogs, cats, and their fleas, respectively, at Clinvet Research Centre, allowing further studies (Table 3).
DNA sequencing, RFLP, and hydrolysis probe genotyping of D. caninum
Primer pairs DC28S-1F and DC28S-1R [3] were used to amplify part of the 28S rDNA region from the genome of D. caninum. PCR products were subjected to Sanger sequencing and sequence assembly of both strands were performed using Geneious assembler (Geneious 8.0.5). All PCR products obtained from Beugnet et al. [3] (Table 1) were subjected to RFLP analysis. PCR product (2 μl) was subjected to direct digestion using 10 units StuI restriction enzyme (New England Biolabs) in a final volume of 20 μl for 1 hour at 37°C. The complete digestion mixture was electrophoretically separated using a 2% (m/v) TAE agarose gel at 6 V/cm for 1 hour. All appropriate controls were included.
The hydrolysis probe qPCR assays to discriminate between the two genotypes were based on a qPCR assay with the following setup. Primers DC28S-1F and DC28S-1R [3] were added to a final concentration of 900 nM each. Probes D_caninum dog (FAM-GTGTGTGCACAGTC-NFQ-MGB) and D_caninum cat (VIC-CCTGTGTGTACAGTCG-NFQ-MGB) were added to a final concentration of 200 nm each. qPCR was performed using a QuantStudio 6 instrument fitted with a 384-well block in a final reaction volume of 10 μl using SsoAdvanced™ Universal Probes Supermix (Bio-Rad) under the following cycling conditions: initial denaturation of 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Pre-and post-read stages at 25°C for 30 sec were included. Analysis was performed using the QuantStudio 6 Real-time PCR system Software. All appropriate controls were included.
Non-invasive field sampling of animals: detection and genotyping of D. caninum
Ninety-nine dogs were sampled in villages under field conditions in the area surrounding Bloemfontein (Free State, Republic of South Africa). The sampling procedure entailed the gentle swabbing of the anal region (including surrounding hair) with a sterile cotton swab. Swabs were collected from dogs living with their owners and stored at room temperature until processing for DNA isolation using the GeneJet Genomic DNA isolation kit (Thermo Fisher Scientific), according to the manufacturer’s recommendation for tissue samples. PCR based detection of D. caninum DNA obtained from 99 anal swabs from dogs resulted in successful amplification of the target region.
During the study, the anal area and hair surrounding it, as well as the sleeping area of the dog were checked for signs of proglottids. Proglottids were expelled by 12 animals at the time of swabbing. The swabs were subjected to PCR analyses, and if positive for D. caninum DNA (Table 3), the dogs were individually placed in kennels at Clinvet and screened for signs of proglottids.
Targeted nuclear DNA amplification and sequencing
Primers WormA (5’-GCGAATGGCTCATTAAATCAG-3’) and WormB (5’- CTTGTTACGACTTTTACTTCC-3’) [11] were used to amplify the 18S ribosomal DNA region from the proglottids of feline (R166 and #1431) and canine origin (CV_ref). The complete 18S region was sequenced using a primer walking strategy. Sequence alignments were performed using the MAFFT plugin in Geneious 8.0.5. Molecular phylogenetic analyses using Bayesian inference and Neighbour-Joining were performed according to Nakao et al. [14].
Primers DC28S-3F (5’-GTGGTCAGGCCTACAGGAGTCGG-3’ based on AF023120) and Universal 28S-1R (5’-CCTGTCTCACGACGGTCTAAACCCAG-3’ based on Hymenolepis diminuta 28S rDNA sequence, AY157181) were used to amplify approx. 2.4 kb of the 28S rDNA region, followed by primer walking to sequence the complete region on both strands.
Sequencing of the mitochondrial DNA
Primer pairs DcMitoUni-1F (5’-GGGCTTGTTTGAATGGTTTGACAAGATAATTTG-3’) and DcMitoUni-1R (5’-CACTTGCTGCCAAACCATTTTAGTTAAAAAACTAAG-3’) were based on the sequence D. caninum partial mt DNA sequence isolated from spotted hyena (Accession number: KF202097) (as published by East et al. [7]).
The 842 bp PCR product was amplified using total DNA isolated from R166 (obtained from the École Nationale Vétérinaire de Maisons-Alfort) and was sequenced using Sanger methodology. The above mentioned primer pairs were reverse complemented, resulting in primers pair DcMitoUni-INV-1F (5- CTTAGTTTTTTAACTAAAATGGTTTGGCAGCAAGTG-3’) and DcMitoUni-INV-1R (5’- CAAATTATCTTGTCAAACCATTCAAACAAGCCC). These primers were used at 400 nM final concentration to amplify the remainder of the mt DNA genome using LongAmp® Taq DNA Polymerase (NEB) using 50 ng total DNA as template. Thermal cycling entailed 94°C for 5 min followed by 40 cycles of 94°C for 30 sec, 59°C for 30 sec, 65°C for 12 min. The thermal cycling was concluded with a final extension of 10 min at 65°C. Purified PCR product of approx. 12 500 bp was submitted to Inqaba Biotec (South Africa) for next generation sequencing making use of Illumina’s Miseq instrument and 2 × 250 bp reads using the MiSeq Reagent Kit v2. Primers F_mtDNA-F (TTCTTGAAGTTTGTCTGTCTGTTT) and F_mtDNA-R (AAGCAGCACATAGACTTAGCTT) were used to amplify 1126 bp, including the DcMitoUni-INV-1F and DcMitoUni-INV-1R primer binding sites and the PCR product was subjected to Sanger sequencing.
Mitochondrial genome sequence analysis
Illumina Miseq and Sanger sequencing data were assembled, after quality trimming, using the Geneious assembler (Geneious 8.0.5). Contigs were mapped to the only available D. caninum mt DNA genome (AB732959), using Geneious assembler. Annotation and similarity percentages of the D. caninum R166 mt genome was performed using the Geneious Live annotate and predict function, making use of AB732959 as reference as well as submitting the final completed mt genome sequence to MITOS [2] for annotation. Geneious Live annotate and predict employs a BLAST-like algorithm that includes translation search where sequences are translated in all six frames and compared to the reference. Results from both automatic annotations were in agreement and were also manually checked.
The 12 protein-coding genes found in the sequenced and annotated mt genome of D. caninum R166 were translated using translation table 9 representing the echinoderm and flatworm mt code. The amino acid sequences were concatenated and compared to the concatenated amino acid sequences obtained from 53 mt genomes from Anoplocephalidae, Dipylidiidae, Hymenolepididae, Paruterinidae and Taeniidae using MAFFT and resulted in an 80.6% BLOSUM62 pairwise similarity after removal of all gaps in the alignment. Maximum Likelihood and Bayesian Inferences analysis were used to construct trees as described by Guo [9] using Schistosoma japonicum derived data as the outgroup.
The 12S rDNA sequence from the newly generated D. caninum feline genotype mt sequence was compared to D. caninum 12S rDNA sequences recently deposited and analyzed by Low et al. [13] using the Mr Bayes (HKY85 substitution model; 1 000 000 chain length and 25% burn-in length) and Schistocephalus solidus as the outgroup.
Results
Sequencing of the 28S and 18S ribosomal DNA regions.
DNA sequence analysis of a 655 bp region of the 28S ribosomal DNA region used in the D. caninum detection PCR (see Table 4) resulted in the identification of two unique groups when these DNA sequences were compared to the GenBank reference sequence (Table 5). One group exhibited 99.5% DNA sequence identity towards the published reference sequence (AF023120) and 100% DNA sequence identity towards the canine derived D. caninum isolated at Clinvet (MH040832), whereas the other group (i.e. “feline genotype”) exhibited a 93.5% sequence identity towards the published reference sequence. As described below in detail, the “100% identity group” DNA isolates were almost all of dog and dog flea origin, whereas the “93.5% identity group” DNA extracts almost all came from cats and fleas collected on cats.
No intra-group differences in DNA sequencing could be observed for any of the samples analyzed using the 655 bp PCR product. Sequence submission to GenBank was under accession numbers indicated in Table 5.
The new “93.5% identity group” corresponds to the feline associated D. caninum samples from the USA, New Zealand, Europe, China and South Africa [with no intrinsic variation related to geographical origin (see Discriminant RFLP analysis below)]. For the clarity of the analysis, we propose to name it “D. caninum feline genotype”.
The “100% identity group” exhibited no intrinsic variation related to geographical origin between all samples from Europe and South Africa. It corresponds to the DNA extracts from infected dogs or fleas collected on dogs (see discriminant RFLP analysis below); therefore we propose to name it “D. caninum canine genotype”.
Amplification and sequence analysis of approx. 2.4 kb 28S rDNA region were performed for two of the “D. caninum feline genotype” representatives and no sequence difference was detected with the 3 ambiguous nucleotides identical between two feline genotypes. The D. caninum canine genotype yielded a sequence with 28 ambiguous nucleotides detected. In spite of the ambiguous nucleotides, a non-ambiguous DNA sequence comparison resulted in differences of 2% (with a 3.1% difference when ambiguous nucleotides were included) between the “D. caninum feline genotypes” (MH045482; MH045483) and the “D. caninum canine genotype” (MH045484) for the 2437 bp compared, with a 12 bp and a 4 bp insertion/deletion event detected.
Amplification of the 18S rDNA region resulted in the amplification of 2.4 kb DNA fragments. 18S rDNA sequence analysis performed on two “D. caninum feline genotype” (MG582181, MG582183) representatives revealed 2.7% differences with the canine genotype (MG582184) with 2 conserved 6 bp and one 8 bp insertions/deletions detected in the feline sequence when compared to the canine sequence (Figure 1). Pairwise sequence comparison also indicate that the D. caninum canine and feline genotypes share sequence identity to a level that can be observed for different species of cestodes when comparing 18S rDNA sequences (Table 6). Phylogenetic analysis based on the 18S rDNA sequences were conducted using sample sets from previous studies [14] and analysis of the sequence identity and patristic distance between the feline genotype, the canine genotype and other cestodes indicated that the sequence identity and patristic distance between the D. caninum feline genotypes and the D. caninum canine genotype was larger than the patristic distance observed between Taenia serialis and Taenia modoquae; Taenia saginata and Taenia asiatica; Taenia martis and Taenia twitchelli as well as between Echinococcus canadensis and Echinococcus ortleppi (Table 6). This suggests that the two D. caninum genotypes are genetically (based on the 18S rDNA sequence data) more distant from a common ancestor than the different pairs of cestode species mentioned above.
Percentage DNA sequence identity obtained from approximately 650 bp PCR product in D. caninum positive samples.
Sample detail table providing GenBank accession numbers.
 |
Figure 1 Conserved insertion/deletion events (indicated by −) present in the 18S rDNA feline genotype representing feline associated D. caninum (bottom sequence) when compared to the canine genotype (second sequence from the bottom) representing the canine associated D. caninum. |
18S rDNA sequence identity and patristic distance observed between the D. caninum feline and canine genotypes and different cestodes.
RFLP and hydrolysis probe genotyping assays.
Sequence data from 28S rDNA region targeted for the PCR revealed the presence of conserved nucleotide differences when comparing the two genotypes. These conserved differences allowed the design of an RFLP assay using the restriction enzyme StuI to generate a product that would allow direct discrimination between the two groups. The StuI recognition occurs twice in the feline genotype and will result in the fragmentation of the 653 bp feline PCR product into 404 bp, 223 bp and 26 bp, and occurs only once in the reference canine genotype, yielding 629 bp and 26 bp fragments from the 655 bp PCR canine product, thereby allowing a clear discrimination between the two identified groups. RFLP analysis was also performed on the 57 sequence verified samples to validate the RFLP assay for accuracy. All results were in concordance.
D. caninum PCR positive samples (n = 192) from the previous study [3] (Table 1) were subjected to this discriminant RFLP analysis. Results indicated that 98.4% of D. caninum infected C. felis fleas collected from dogs were genotyped as the canine genotype, while only 1.6% of the positive fleas were genotyped as belonging to the feline genotype. 100% of infected Pulex irritans and C. canis fleas were harbouring the canine genotype of D. caninum.
RFLP analysis also showed that 90.7% of infected fleas from cats were genotyped as belonging to the feline genotype with only 9.3% of D. caninum infected fleas from cats exhibiting the canine genotype.
A hydrolysis probe genotyping assay, based on the validated PCR screening assay, was developed to allow for high throughput genotyping of the two identified groups. A 2 bp insertion/deletion present in the 653/655 bp PCR target region served as a target site for the genotyping probes. The hydrolysis probe genotyping assay was validated on samples that were subjected to Sanger sequencing and RFLP analyses. All three assays were 100% in agreement. The hydrolysis probe genotyping assays were also able to discriminate between homozygous for the “canine genotype”, homozygous for the “feline genotype” and an artificial heterozygous mix of DNA from both genotypes. This technique could serve to identify the possibility or not of hybridization between the two genotypes in definitive hosts, as cats and dogs are often present together in a same household [4].
Identification and isolation of a Dipylidium canine genotype strain at Clinvet International
In order to collect a local canine genotype of D. caninum and to assess the possibility of using the PCR technique on anal swabs, 38 flea-infested dogs living in the village near ClinVet were assessed through a veterinary consultation (Table 3). Twelve dogs expelling Dipylidium proglottids were placed in kennels at ClinVet. Anal swabbing was conducted on all of these dogs and the swabs were subjected to PCR analyses. All 12 animals that were diagnosed infected in the field were also diagnosed positive by PCR on swabs. The 26 other dogs classified as not expelling proglottids during field direct examination, were found positive by swab PCR. These 26 dogs started to expel proglottids later in the next few days. The proglottids were genotyped to confirm the canine genotype, and were then pooled to start breeding the canine genotype colony at Clinvet by passing through fleas and dogs.
Mitochondrial DNA amplification of sequence analysis.
Total DNA extract from adult D. caninum R166 isolate (representing the feline genotype based on 18S and 28S sequence analysis and genotyping) was used to amplify the complete mt genome in two fragments. The partial feline genotype 842 bp fragment revealed a 99% identity towards KF202097, the partial sequence obtained from the spotted hyena [7], and only an 89% identity towards the completed D. caninum mt genome from the reference group, i.e. NC_021145; “canine genotype”.
The remainder of the mt genome was amplified by simply reverse complementing both primers used to amplify the 842 bp fragment. The approx. 13 kb fragment was sequenced using Illumina chemistry resulting in 215 263 reads with a mean read length of 196 bp resulting in > 3000X coverage of the expected mt genome. Mapping the reads to the D. caninum mt genome (AB732959) resulted in assembly of the complete mt genome from D. caninum R166 (representing the “cat genotype”) with an mt genome size of 13 598 bp (Figure 2; GenBank accession number: MG587892). Nucleotide analysis revealed an overall base composition of 22.3% A, 8.8% C, 19.7% G and 49.1% T, resulting in a low GC content of 28.5%.
Direct DNA comparison of the complete mtDNA genomes of D. caninum R166 (“feline genotype” MG587892) and the reference D. caninum mtDNA genome (AB732959 and NC_021145; “canine genotype”) indicate only a 78.7% identity on the DNA level. The DNA sequences of AB732959 and NC_021145 are identical, but they are annotated differently.
Analyses of the CDS, rRNA and tRNA regions (36 in total) with the specific associated identities between the two mt genomes are represented in Table 7. The ATP8 coding gene, present in mammalian mt genomes, could not be observed in the D. caninum mt genome, which is in agreement with published tapeworm mt genomes.
No STOP codon could be detected for the COX1 protein-coding region from MG587892 and differently annotated COX1 encoding sequences are reported for AB732959 and NC_021145. Both COX1 annotations for the “canine genotype” mt DNA are under review from GenBank (GenBank email communication).
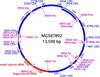 |
Figure 2 Graphical representation of the complete mitochondrial genome of D. caninum R166 (MG587892) including the organization and direction of 36 genes within the mitochondrial genome. |
Results following analysis of the CDS, rRNA and tRNA regions (36 in total) with the specific associated identities between the two mitochondrial genomes.
Mitochondrial phylogenetic analysis.
Mitochondrial genomes used by Guo [9], including any updated and additional genomic DNA sequences, were downloaded from GenBank and used in all subsequent analyses. Concatenation of the 12 protein-coding genes from D. caninum feline genotype (R166 adult tapeworm isolate), canine genotype, and those of 52 other tapeworms were subjected to multiple alignment followed by maximum likelihood and Bayesian inference analysis (average standard deviation of split frequencies was below 0.005) using Schistosoma japonicum as the outgroup. Both analysis methods exhibited the same topology and confidence and the tree obtained from the Bayesian inference is shown in Figure 3.
Protein identity analysis (based on the concatenated mt protein sequences) between the “canine genotype” and the “feline genotype” of D. caninum mt genomes revealed only an 81.8% identity between the two genotypes, which is 17.2% lower than the average protein identity calculated for the other tapeworm genotypes available from GenBank. The patristic distance of 0.33 between the two genotypes is more than 7-fold the average patristic distance of the other sequence genotypes used in the analysis. The patristic distance between the D. caninum canine and feline genotypes is larger than the patristic distance between any of the Echinococcus spp., Spirometra spp. and Diphyllobothrium spp. used in the analysis. This analysis provides genetic support that the shared identity and the patristic distance scores observed between the dog and the cat genotypes identify two different Dipylidium species, and not only intra-species genotypes.
 |
Figure 3 Tree obtained after concatenation of the 12 protein-coding genes from D. caninum R166 and after those of 52 other tapeworms were subjected to multiple alignment, followed by maximum likelihood and Bayesian inference analysis (average standard deviation of split frequencies was below 0.005), using Schistosoma japonicum as the outgroup. |
Discussion
The molecular characterization of D. caninum isolates collected from dogs, cats, and in infected fleas collected either from dogs or cats allowed the identification of two distinct genotypes that clearly differ from each other.
East et al., 2013, collected D. caninum proglottids from six spotted hyena [7]. Initial PCR amplification and sequencing of the 314 bp fragment indicated identical sequence data for the partial 12S mt rDNA region from all six proglottids. Comparison of 314 bp sequence data with two published D. caninum sequences revealed a high (99%) similarity to one sequence from Europe (accession number L49460.1) but a considerably lower similarity (89%) to one sequence from Asia (accession number AB031362.1). They selected one of the six samples and PCR amplified and sequenced 1176 bp of the 12S mt rDNA (accession number KF202097). Comparison of this sequence to a similar fragment from D. caninum, again revealed a relative low similarity (89%). Pairwise sequence comparison between the sequences of East et al., 2013 and our complete mt sequence of the D. caninum feline genotype (MG587892), revealed a 99.1% identity between the D. caninum isolated from the hyena (KF202097) and the D. caninum feline genotype (MG567892) isolated from a cat. When comparing these sequences to the mt genome of the D. caninum dog genotype, there is only an 88.5% and an 88.8% identity, respectively. This confirms that the Dipylidium isolate from hyena belongs to the “feline genotype”.
More recently, Low et al., 2017, collected ectoparasites on dogs and cats in Malaysia [13]. In this study, C. felis (92 specimens) and Felicola subrostratus (30 specimens) were collected from 20 cats, whereas C. orientis (26 specimens) and Rhipicephalus sanguineus sensu lato (120 specimens) were collected from 29 dogs. PCR utilizing the primers we published in 2014 [3] was performed to amplify the partial 28S rDNA gene region of D. caninum. They found 2% of cat fleas and 10% of cat lice infected by D. caninum. They indicated that the representative 28S rRNA sequence isolated from their flea and louse specimens (accession no. KY751956) demonstrated 95% sequence similarity with that of D. caninum (accession no. AF023120), and they suggested the existence of a second distinct species from the one available in GenBank. This 5% divergence of the approx. 650 bp region of the 28S rDNA is consistent with data reported in this study. PCR amplification and sequencing of the partial 12S rDNA gene region indicated that the 12S rDNA sequences (accession no. KY751955) were clustered together with those adult specimens isolated from red fox (accession no. L49460) and spotted hyena (accession no. KF202097). They found a high level of genetic distance (9.59%) and concluded on the existence of two clades, with a genetic divergence comparable with that of species pairs in their relatives from the genus Echinococcus (1.31–10.06%) [13].
We compared the 12S mt rDNA sequence of MG587892 to D. caninum 12S mt rDNA sequences used by Low et al. [13], and Bayesian Inference phylogenetic analysis clearly clustered the D. caninum feline genotype with their D. caninum isolated from cat fleas and cat lice collected from cats (Figure 4). This clustering with the D. caninum sequence data independently obtained by Low et al. [13] confirms the clear association of the D. caninum feline genotype with cats. The hypothesis drawn by Low et al. [13] on the existence of two clades is confirmed by the presented work and corresponds to the canine and feline genotypes described in this paper.
These two genotypes are not related to geographical origin as they were found by several authors [7,13] and in the present study in Dipylidium sp. from all continents (i.e. North America, Europe, Asia, and Africa), Tables 1–Tables 1 to 3, but clearly to their host origin, dogs or cats (and hyena). Nevertheless, a small proportion (from 2 to 10%) of D. caninum DNA extracted from cats or C. felis fleas collected from cats, or extracted from dogs or C. felis fleas collected from dogs, belong to the other genotype. The specificity therefore does not appear to be absolute and should be studied by experimental infections in both dogs and cats (Beugnet et al., 2018, Part 2, [4]). The common presence of both cats and dogs in the same households, being infested by the same fleas (i.e. C. felis), may explain the infection of cats and dogs by both genotypes, but the different observed prevalences suggest biological adaptation. On the other hand, in C. canis and P. irritans fleas, being more specific to dogs, 100% of the infected fleas were found to harbour the canine genotype of D. caninum (Table 1).
A comparison of biological development and host preference should confirm the genetic observations (Beugnet et al., 2018, [4]). The genetic differences observed in this analysis, which show a greater distance to what is known between different species of Taenia or Echinococcus, make it possible to suggest the existence of two distinct Dipylidium species, which will have to be confirmed or disproved.
 |
Figure 4 12S mt rDNA tree obtained for D. caninum sequences from GenBank using Schistocephalus solidus as the outgroup. |
Conflicts of interest
The authors declare that they have no conflicts of interest in relation to this article.
Acknowledgements
The authors would like to thank the research personnel from both Clinomics and Clinvet International (Pty) Ltd for their assistance in conduct of the research. The study was funded by Merial SAS, France. There were no conflicts of interest.
Special thanks to Prof. M. Lappin, North Carolina State University, USA; Prof. Piyanan Taweethavonsawat, Chulalongkorn University, Bangkok, Thailand; Prof. Elias Papadopoulos, Thessaloniki Veterinary Faculty, Greece; Prof Smaro Sotiraki, Hellenic Agricultural Organisation, Greece; and Dr Maureen Forsyth, Auckland, New Zealand, for the collection and shipment of fleas and Dipylidium sp. tapeworms.
References
- Allsop BA, Jones A, Allsop MTEP, Newton SD, Macpherson CNL. 1987. Interspecific characterization of several taeniid cestodes by isoenzyme analysis using iselectric focusing in agarose. Parasitology, 95, 593-601. [CrossRef] [PubMed] [Google Scholar]
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution, 69 (2), 313-319. [CrossRef] [PubMed] [Google Scholar]
- Beugnet F, Labuschagne M, Fourie J, Jacques G, Farkas R, Cozma V, Halos L, Hellmann K, Knaus M, Rehbein S. 2014. Occurrence of Dipylidium caninum in fleas from client-owned cats and dogs in Europe using a new PCR detection assay. Veterinary Parasitology, 205, 300-306. [CrossRef] [PubMed] [Google Scholar]
- Beugnet F, Labuschagne M, de Vos C, Crafford D, Fourie J. 2018. Analysis of Dipylidium caninum tapeworms from dogs and cats, or their respective fleas. Part 2. Distinct canine and feline host association with two Dipylidium caninum different genotypes. Parasite, 25, 31. [CrossRef] [EDP Sciences] [PubMed] [Google Scholar]
- Chandra S, Forsyth M, Lawrence AL, Emery D, Šlapeta J. 2017. Cat fleas (Ctenocephalides felis) from cats and dogs in New Zealand: Molecular characterisation, presence of Rickettsia felis and Bartonella clarridgeiae and comparison with Australia. Veterinary Parasitology, 234, 25-30. [CrossRef] [PubMed] [Google Scholar]
- Crafford D, Kok D. 2013. Die problematiek verbonde aan die identifikasie van Taenia spp. in honde (Canis familiaris), jakkalse (Canis mesomelas) en rooikatte (Caracal caracal) in Suid-Afrika. LitNet Akademies, 10 (2), 109-139. [Google Scholar]
- East ML, Kurze C, Wilhelm K, Benhaiem S, Hofer H. 2013. Factors influencing Dipylidium sp. infection in a free-ranging social carnivore, the spotted hyaena (Crocuta crocuta). International Journal for Parasitology: Parasites and Wildlife, 2, 257-265. [CrossRef] [Google Scholar]
- Gates MC, Nolan TJ. 2009. Endoparasite prevalence and recurrence across different age groups of dogs and cats. Veterinary Parasitology, 10, 153-158. [CrossRef] [PubMed] [Google Scholar]
- Guo A. 2016. The complete mitochondrial genome of the tapeworm Cladotaenia vulturi (Cestoda: Paruterinidae): gene arrangement and phylogenetic relationships with other cestodes. Parasites & Vectors, 9, 475. [CrossRef] [PubMed] [Google Scholar]
- Lahmar S, Boufana B, Ben Boubaker S, Landolsi F. 2014. Intestinal helminths of golden jackals and red foxes from Tunisia. Veterinary Parasitology, 204, 297-303. [CrossRef] [Google Scholar]
- Littlewood DTJ, Olson PD. 2001. Small subunit rDNA and the phylum Platyhelminthes: signal, noise, conflict and compromise, in Interrelationships of the Platyhelminthes, Littlewood DTJ, Bray RA, Editors. Taylor and Francis: London. 262-278. [Google Scholar]
- Loos-Frank B. 2000. An up-date of Verster’s (1969) ’Taxonomic revision of the genus Taenia Linnaeus’ (Cestoda) in table format. Systematic Parasitology, 45, 155-183. [CrossRef] [PubMed] [Google Scholar]
- Low VL, Prakash BK, Tan TK, Sofian-Azirun M, Anwar FHK, Vinnie-Siow WY, AbuBakar S. 2017. Pathogens in ectoparasites from free-ranging animals: Infection with Rickettsia asembonensis in ticks, and a potentially new species of Dipylidium in fleas and lice. Veterinary Parasitology, 245, 102-105. [CrossRef] [PubMed] [Google Scholar]
- Nakao M, Lavikainen A, Iwaki T, Haukisalmi V, Konyaev S, Oku Y, Okamoto M, Ito A. 2013. Molecular phylogeny of the genus Taenia (Cestoda: Taeniidae): proposals for the resurrection of Hydatigera Lamarck, 1816 and the creation of a new genus Versteria. International Journal for Parasitology, 43, 427-437. [CrossRef] [PubMed] [Google Scholar]
- Sahin I, Köz S, Atambay M, Kayabas U, Piskin T, Unal B. 2015. A rare cause of diarrhea in a kidney transplant recipient: Dipylidium caninum. Transplantation Proceedings, 47, 2243-2244. [CrossRef] [PubMed] [Google Scholar]
- Venard CE. 1938. Morphology, bionomics, and taxonomy of the cestode Dipylidium caninum. Annals of the New York Academy of Science, 37, 273-328. [CrossRef] [Google Scholar]
- Verster A. 1969. A taxonomic revision of the genus Taenia Linnaeus, 1758, s. str. Onderstepoort. Journal of Veterinary Research, 36 (1), 3-58. [Google Scholar]
- Witenberg G. 1932. On the cestode subfamily Dipylidiinae Stiles. Zeitschrift für Parasitenkunde, 4, 541-584. [CrossRef] [Google Scholar]
- Zhang L, Hu M, Jones A, Allsopp BA, Beveridge I, Schindler AR, Gasser RB. 2007. Characterization of Teania madoquae and Taenia regis from carnivores in Kenya using genetic markers in nuclear and mitochondrial DNA, and their relationship with other selected teaniids. Molecular and Cellular Probes, 21, 379-385. [CrossRef] [PubMed] [Google Scholar]
Cite this article as: Labuschagne M, Beugnet F, Rehbein S, Guillot J, Fourie J, Crafford D. 2018. Analysis of Dipylidium caninum tapeworms from dogs and cats, or their respective fleas. Parasite 25, 30
All Tables
Details on two distinct 28S rDNA sequence variants (“canine” and “feline”) defined from fleas infected by Dipylidium caninum, obtained from PCR products collected by Beugnet et al. [3] from Europe and South Africa.
Details on two distinct 28S rDNA sequence variants (“canine” and “feline”) defined from fleas infected by Dipylidium caninum, received from the United States of America.
Details on two distinct 28S rDNA sequence variants (“canine” and “feline”) defined from non-invasive anal swabs collected from cats and dogs infected by Dipylidium caninum in South Africa.
Percentage DNA sequence identity obtained from approximately 650 bp PCR product in D. caninum positive samples.
18S rDNA sequence identity and patristic distance observed between the D. caninum feline and canine genotypes and different cestodes.
Results following analysis of the CDS, rRNA and tRNA regions (36 in total) with the specific associated identities between the two mitochondrial genomes.
All Figures
|
Figure 1 Conserved insertion/deletion events (indicated by −) present in the 18S rDNA feline genotype representing feline associated D. caninum (bottom sequence) when compared to the canine genotype (second sequence from the bottom) representing the canine associated D. caninum. |
| In the text | |
|
Figure 2 Graphical representation of the complete mitochondrial genome of D. caninum R166 (MG587892) including the organization and direction of 36 genes within the mitochondrial genome. |
| In the text | |
|
Figure 3 Tree obtained after concatenation of the 12 protein-coding genes from D. caninum R166 and after those of 52 other tapeworms were subjected to multiple alignment, followed by maximum likelihood and Bayesian inference analysis (average standard deviation of split frequencies was below 0.005), using Schistosoma japonicum as the outgroup. |
| In the text | |
|
Figure 4 12S mt rDNA tree obtained for D. caninum sequences from GenBank using Schistocephalus solidus as the outgroup. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.