| Issue |
Parasite
Volume 30, 2023
|
|
|---|---|---|
| Article Number | 58 | |
| Number of page(s) | 13 | |
| DOI | https://doi.org/10.1051/parasite/2023057 | |
| Published online | 12 December 2023 | |
Research Article
Microbial diversity of ticks and a novel typhus group Rickettsia species (Rickettsiales bacterium Ac37b) in Inner Mongolia, China
Diversité microbienne des tiques et nouvelle espèce de Rickettsia du groupe du typhus (bactérie Rickettsiales Ac37b) en Mongolie intérieure, Chine
1
Graduate School, Inner Mongolia Medical University, Hohhot 010059, Inner Mongolia, China
2
School of Basic Medicine, Inner Mongolia Medical University, Hohhot 010110, Inner Mongolia, China
3
First Hospital of Jilin University, Changchun 130021, China
4
Hulunbuir Mental Health Center, Hulunbuir 022150, Inner Mongolia, China
5
Beijing Guoke Biotechnology Co., Ltd, 102200 Beijing, China
6
Department of Infection Control, Second Affiliated Hospital of Inner Mongolia Medical University, Hohhot, Inner Mongolia Autonomous Region 010000, China
7
Inner Mongolia Academy of Agricultural & Animal Husbandry Science, Hohhot 010031, Inner Mongolia, China
* Corresponding authors: This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
1
August
2023
Accepted:
20
November
2023
Abstract
Ticks can carry multiple pathogens, and Inner Mongolia’s animal husbandry provides excellent environmental conditions for ticks. This study characterized the microbiome of ticks from different geographical locations in Inner Mongolia; 905 Dermacentor nuttalli and 36 Ixodes persulcatus were collected from sheep in three main pasture areas and from bushes within the forested area. Mixed DNA samples were prepared from three specimens from each region and tick species. Microbial diversity was analyzed by 16S rRNA sequencing, and α and β diversity were determined. The predominant bacterial genera were Rickettsia (54.60%), including Rickettsiales bacterium Ac37b (19.33%) and other Rickettsia (35.27%), Arsenophonus (11.21%), Candidatus Lariskella (10.84%), and Acinetobacter (7.17%). Rickettsia bellii was identified in I. persulcatus, while Rickettsiales bacterium Ac37b was found in D. nuttalli from Ordos and Chifeng. Potential Rickettsia and Anaplasma coinfections were observed in the Ordos region. Tick microbial diversity analysis in Inner Mongolia suggests that sheep at the sampling sites were exposed to multiple pathogens.
Résumé
Les tiques peuvent être porteuses de plusieurs agents pathogènes et l’élevage en Mongolie intérieure offre d’excellentes conditions environnementales pour les tiques. Cette étude a caractérisé le microbiome des tiques de différentes zones géographiques de Mongolie intérieure; 905 Dermacentor nuttalli et 36 Ixodes persulcatus ont été collectés sur des moutons dans trois principales zones de pâturage et dans des buissons de la zone forestière. Des échantillons d’ADN mixtes ont été préparés à partir de trois spécimens de chaque région et espèce de tique. La diversité microbienne a été analysée par séquençage de l’ARNr 16S et la diversité α et β a été déterminée. Les genres bactériens prédominants étaient les Rickettsia (54,60 %), dont la bactérie Rickettsiales Ac37b (19,33 %) et d’autres Rickettsia (35,27 %), Arsenophonus (11,21 %), Candidatus Lariskella (10,84 %) et Acinetobacter (7,17 %). Rickettsia bellii a été identifiée chez I. persulcatus, tandis que la bactérie Rickettsiales Ac37b a été trouvée chez D. nuttalli d’Ordos et Chifeng. Des co-infections potentielles à Rickettsia et Anaplasma ont été observées dans la région d’Ordos. L’analyse de la diversité microbienne des tiques en Mongolie intérieure montre que les moutons présents sur les sites d’échantillonnage sont exposés à plusieurs agents pathogènes.
Key words: Inner Mongolia / Dermacentor nuttalli / Ixodes persulcatus / Microbial diversity / Rickettsiales bacterium Ac37b
Edited by Jean-Lou Justine
Equal contributors
© S. Su et al., published by EDP Sciences, 2023
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Ticks are arachnids (Arachnida: Acari: Ixodida) that parasitize birds, reptiles, amphibians, and terrestrial mammals, including humans. Ticks are bloodsucking at all developmental stages. When biting their hosts, ticks can be vectors for the transmission of various pathogens and cause irritation, anemia, and local or systemic hypersensitivity [37]. Ticks are second only to mosquitos as human disease vectors throughout the world [8, 19, 68]. They can transmit numerous pathogens, such as viruses, bacteria, and protozoa [32]. Rickettsia are vertically transmitted symbionts in invertebrates and pathogens in vertebrates. Tick-borne rickettsioses are caused by obligatory intracellular bacteria from the spotted fever group of the genus Rickettsia [43]. In the past few decades, tick-borne Rickettsia infections have been reported worldwide, most of which are widely distributed in tropical and subtropical areas, and the scope of infection is expanding. Although some Rickettsiales isolates are not associated with pathogenicity in vertebrates, thirteen tick-borne Rickettsiales pathogens have been reported to cause human disease in China, including seven spotted fever group Rickettsia (SFGR), two species of Ehrlichia, and four other species in the family Anaplasmataceae [12, 14]. Ticks are found all across China; however, as their habitat environment greatly affects where they are located, ticks are more frequently found in forested areas [3, 21, 22, 50, 62, 70]. Depending on the pathogen, the severity of tick-borne disease (TBD) can even be life-threatening. As human populations have grown, their interactions with the wild have increased. Additionally, human exposure to ticks carrying pathogens has greatly increased [55].
The transmission process of TBD is influenced by many factors, including pathogens, vectors, potential hosts, the environment, and human behavior. In addition, TBDs often benefit from human population mobility, animal migration, and global logistics. In particular, global logistics increase the possibility of tick-borne pathogens spreading across borders [65]. Worldwide, TBDs, such as tick-borne encephalitis, Crimean-Congolese hemorrhagic fever, and Rocky Mountain spotted fever, have posed new threats to public health worldwide, and the incidence of TBD is increasing at an alarming rate [34, 46]. The network of authorities in the United States reported nearly 650,000 cases of vector-borne diseases from 2004 to 2016. More than 75% of these cases were TBDs [48]. From 2016 to 2019, more than 200,000 cases of nine TBDs were reported to the Centers for Disease Control and Prevention (CDC). However, due to underreporting, the number of cases may be higher [48]. In addition, in mainland China, 33 new tick-borne diseases have been identified since the early 1980s, indicating a significant public health danger [64].
China is a large country with complicated topographical features, a varied climate, and several types of ticks. Therefore, TBDs are prevalent in most areas of China, seriously affecting human health [72]. Zhao et al. found that D. nuttalli is a tick species that carries many different tick-borne pathogens in China. According to a prediction model [71], the habitat suitable for 19 tick species was 14–476% larger than the geographical area where these species are currently found, indicating that there are still serious deficiencies in our knowledge of tick distribution. Due to large pasture areas and extensive animal husbandry, Inner Mongolia provides an excellent habitat for ticks, and the incidence of zoonosis is often very high [66]. In 2005, Jia et al. [17] reported human cases of Rickettsia raoultii in northeast China for the first time.
Ticks and TBDs have a substantial impact on the economy and human life in Inner Mongolia. Nevertheless, the community structure and diversity of microbial communities on ticks parasitizing sheep in various regions of Inner Mongolia have not been thoroughly studied thus far. The microbial communities of ticks are influenced by many factors, including geographical area, feeding status, blood meal source, and developmental stage [25, 49, 63].
Current techniques for 16S ribosomal RNA (rRNA) gene sequence analysis, based on typical clustering thresholds of operational taxonomic units (OTUs), are insufficient for accurate taxonomic annotation and for addressing phylogenetic relationships at the species level when only a few hypervariable regions are amplified. Therefore, amplified 16S rRNA gene sequence data of the V3–V4 region can only be explored at the genus level [45]. PacBio full-length 16S rDNA third-generation sequencing technology, however, is more accurate and can be used for reliable species identification [56].
Therefore, we applied this technology to analyze the microbial diversity of ticks collected in four main pastoral areas of Inner Mongolia. The aim of our investigation was to assess the distribution characteristics of tick microbial communities in different geographical locations in Inner Mongolia, providing information for better environmental management.
Materials and methods
Tick collection and sample preparation
In mid-April 2019, adult ticks were collected with tweezers and stored in breathable bottles from bushes in the Arshan Forest area and from sheep pastures in Hulun Buir City (New Barag Right Banner), Chifeng City (Bayan Wendusumu area, Tianshan Town, Alukeerqin Banner), and Ordos City (Chengchuan Town, Otog Front Banner), Inner Mongolia (Fig. 1).
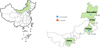 |
Figure 1 Map of tick collection sites in Inner Mongolia, China. |
According to morphological identification based on the literature [15], the 905 ticks from Hulun Buir, Chifeng, and Ordos were all D. nuttalli. However, 36 ticks from bushes in the Arshan Forest area were identified as I. persulcatus. The morphological identification of the ticks was then double checked with molecular biology methods based on the sequences of the species-specific 16S rRNA gene [53, 59]. The 905 D. nuttalli and 36 I. persulcatus were stored at −80 °C until further processing.
DNA extraction
The ticks were disinfected with 75% ethanol, dried on filter paper, washed three times with PBS, and finally dried on filter paper. A total of 941 ticks were divided into 206 sample pools. In each sample pool, 1–20 ticks were placed into a sterile grinding tube. One 5 mm magnetic bead and two 3 mm magnetic beads were added, and the grinding tube was placed into the adapter (cooled to −20 °C in a freezer for 30 min). The adapter was installed into the high-speed tissue homogenizer (Servicebio, Wuhan, China), and the parameters were set to 70 Hz and 180 s. Finally, according to the instructions for the tissue genomic DNA extraction kit (TIANGEN, Beijing, China), all extracted DNA was stored at −20 °C. Three DNA samples (Table 1) were randomly selected from each sample area, and 10 μL of each sample was prepared for microbial diversity sequencing.
Information on tick samples used for bacterial microbiome analysis.
16S rRNA amplicon sequencing
The V1–V9 regions of the bacterial 16S rRNA gene were amplified by PCR (95 °C for 2 min, followed by 27 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 60 s and a final extension at 72 °C for 5 min) using the primers 27F 5′–AGAGTTTGATCCTGGCTCAG–3′ and 1492R 5′–GGTTACCTTGTTACGACTT–3′ [39], where the barcode is an eight-base sequence unique to each sample. PCRs were performed in triplicate in a 20-μL mixture containing 4 μL of 5× FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. Amplicons were extracted from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA), according to the manufacturer’s instructions. SMRTbell libraries were prepared from the amplified DNA by blunt ligation according to the manufacturer’s instructions (Pacific Biosciences). Purified SMRTbell libraries from the pooled and barcoded samples were sequenced on dedicated PacBio Sequel cells using S/P1-C1.2 sequencing chemistry. Replicate 1 of the samples was sequenced using S/P2-C2/5.0 sequencing chemistry. Replicate 2 of the samples was sequenced with a prerelease version of the S/P3-C3/5.0 sequencing chemistry. All amplicon sequencing was performed by Shanghai Biozeron Biotechnology Co., Ltd. (Shanghai, China).
Bioinformatic analysis of 16S rRNA amplicon sequence data
PacBio raw reads were processed using SMRT Link Analysis software version 6.0 to obtain demultiplexed circular consensus sequence (CCS) reads with the following settings: minimum number of passes = 3, minimum predicted accuracy = 0.99. Raw reads were processed through the SMRT Portal to filter sequences for length (<800 or >2500 bp) and quality. Sequences were further filtered by removing barcodes, primer sequences, chimeras and sequences that contained 10 consecutive identical bases. OTUs were clustered with a 97% similarity cutoff using UPARSE (RRID:SCR_005020), and chimeric sequences were identified and removed using UCHIME. The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed with the RDP Classifier [7] (http://rdp.cme.msu.edu/) against the SILVA (RRID:SCR_006423) 16S rRNA database using a confidence threshold of 70% [2]. Subsequently, α and β diversity analyses were run. Rarefaction analysis based on Mothur (RRID:SCR_011947) [51] was used to reveal the Chao, ACE, and Shannon diversity indices. The β diversity analysis was performed using UniFrac (RRID:SCR_014616) [30] to compare the results of the principal coordinates analysis (PCoA) using the community ecology package R-forge (the vegan 2.0 package was used to generate a PCoA figure). One-way analysis of variance (ANOVA) tests were used to assess the statistically significant differences in diversity indices between samples. Differences were considered significant at p < 0.05. Venn diagrams were drawn using the online tool “Draw Venn Diagram” (http://bioinformatics.psb.ugent.be/webtools/Venn) to analyze overlapping and unique OTUs during the treatment processes.
For the identification of biomarkers for highly dimensional colonial bacteria, linear discriminant analysis effect size (LEfSe) was performed [52]. The Kruskal–Wallis sum rank test was applied to examine the changes and dissimilarities among classes followed by linear discriminant analysis (LDA) to determine the size effect of each distinctively abundant taxon [16].
Phylogenetic analyses
Using Mega 7 software, sequences taken from the GenBank database were aligned with the ones obtained, and phylogenetic trees for the 16S rRNA gene were constructed using neighbor-joining (NJ) modeling to confirm the phylogenetic relationships of the pathogens discovered in this study. Bootstrap values for the individual branches of the resulting tree were determined by applying bootstrap analysis with 1,000 replicates. New sequences were deposited in GenBank with accession numbers OQ852069–OQ852073 and OP286852–OP286858.
Results
Identification of ticks
A total of 941 adult hard ticks were identified as D. nuttalli (n = 905) (accession number: OQ852069) and I. persulcatus (n = 36) (accession number: OQ852073) with molecular biology and morphological identifications. Two PCR products from each of the two tick species were randomly selected for sequencing comparison to confirm the morphological identifications, and both 16S rRNA gene sequences obtained were uploaded to the GenBank database.
PacBio sequencing data
A total of 12 samples were sequenced (Table 1). After data screening and deletion, a total of 201,159 reads were generated and classified. The reads of each sample ranged from 11,550 to 21,996. The dilution curve of the Shannon index at the OTU level showed a suitable range for sequencing, and the observed Shannon index accumulation curve also reached a plateau (Fig. 2).
 |
Figure 2 Shannon–Wiener curve. X-axis: amount of sequencing data; Y-axis: corresponding Shannon diversity index. |
Bacterial microbiome composition
A total of 326 OTUs were detected in 12 samples, and the bacterial microbial components were 11 phyla, 15 classes, 38 orders, 62 families, 104 genera, and 141 species. At the genus level (Fig. 3a), the abundance of Rickettsia was the highest at 54.60%, including Rickettsiales bacterium Ac37b (19.33%) and other Rickettsia (35.27%), followed by Arsenophonus (11.21%), Candidatus Lariskella (10.84%), Acinetobacter (7.17%), Cupriavidus (2.39%), and Romboutsia (1.00%). The most abundant Rickettsia were found in D. nuttalli in the Chifeng area, followed by I. persulcatus in the Arshan area, whereas Candidatus Lariskella was found primarily in I. persulcatus in Arshan. At the species level (Fig. 3b), R. raoultii had the highest abundance (23.23%), followed by Rickettsiales bacterium Ac37b (19.33%), Rickettsia sp. (11.94%), Candidatus Lariskella sp. (10.84%), and Arsenophonus sp. (10.32%). Rickettsia raoultii was found in samples of I. persulcatus and the majority of D. nuttalli, except in Ordos. Most Rickettsiales bacterium Ac37b was found in D. nuttalli of Ordos and Chifeng, and was most prevalent in Ordos.
 |
Figure 3 Microbe composition of different regions and species. (a) Top 10 microbial components at the genus level. (b) Top 12 microbial components at the species level. |
Differences in ticks in different areas
There were 180, 182 and 135 OTUs in the samples of D. nuttalli from Hulun Buir, Chifeng and Ordos, respectively. According to the α diversity, the Hulun Buir region showed greater microbial diversity than the other two regions (Fig. 4). LEfSe showed that R. raoultii, Peptostreptococcaceae, and Clostridia played an important role in the Chifeng area. Anaplasma was characteristic of the Ordos formation, while Enterobacterales and Xanthomonadaceae were found at Hulun Buir (Fig. 5). To further distinguish the composition of the microbial community, weighted UniFrac analyses revealed differences regarding the region, as measured by an analysis of similarity (ANOSIM, R = 0.7994, p = 0.011) and visualized by PCoA (Fig. 6). PCoA explained 47.46% (Axis 1) and 28.93% (Axis 2) of the variation, with samples from different regions clustering separately (Fig. 6).
 |
Figure 4 Alpha diversity measures for Dermacentor nuttalli and Ixodes persulcatus in four areas. (a) Shannon’s index. (b) Simpson’s index. |
 |
Figure 5 (a) Clustering tree analysis by linear discriminant analysis effect size (LEfSe). (b) Histogram of LDA analysis. |
 |
Figure 6 PCoA of β-diversity measures for twelve groups. Weighted UniFrac PCoA graph showing PC1, which accounts for 47.46% of variation, and PC2, which accounts for 28.93% of variation. Different colored dots represent different regions and species. |
Differences between Dermacentor nuttalli and Ixodes persulcatus
North of the Arshan region is adjacent to the New Barag Left Banner and to the Ewenki Autonomous Banner of Hulun Buir City, close to the Hulun Buir tick collection point. All I. persulcatus came from the Arshan region. In the Hulun Buir area, only D. nuttalli was found. A total of 123 microbial OTUs were found in ticks, and α diversity indicated that specimens of D. nuttalli contained greater microbial diversity than specimens of I. persulcatus (Fig. 4). Analysis of OTU clusters at the genus level of D. nuttalli and I. persulcatus samples found that their most common bacteria were Acinetobacter, Cutibacterium, Pseudomonas, Ralstonia, Rhodanobacter, Rickettsia and Vibrionimonas (Fig. 7).
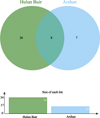 |
Figure 7 Venn diagram for cluster analysis of OTUs between Dermacentor nuttalli and Ixodes persulcatus (genus level). |
Analysis of major bacteria and pathogenic bacteria
We selected bacteria with high abundance and obvious harm to humans at the species level (R. raoultii, Anaplasma and Coxiella) for further analysis (Fig. 8).
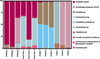 |
Figure 8 The composition of bacteria and pathogenic bacteria with a high abundance. |
Phylogenetic analysis of Rickettsiales bacterium Ac37b
The Rickettsiales bacterium Ac37b (OP286853 and OP286855) sequences were randomly selected to build a phylogenetic tree, named after regions and displayed on the phylogenetic tree with squares and triangles (Fig. 9a). The phylogenetic tree of the 16S rRNA gene showed that Rickettsiales bacterium Ac37b found in Inner Mongolia is in the same branch as the Rickettsiales bacterium Ac37b (CP009217) found in Amblyomma cajennense from Brazil, and their homology is 100% according to sequence alignment analysis using MEGA 7.0 software.
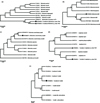 |
Figure 9 Phylogenetic tree of Rickettsiales bacterium Ac37b, Rickettsia bellii, Rickettsia raoultii, Anaplasma and Coxiella in ticks based on neighbor-joining (NJ) modeling; only values higher than 60 were added to the tree branches. (a) Phylogenetic tree of Rickettsiales bacterium Ac37b identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (1320 bp) and triangles (1430 bp). (b) Phylogenetic tree of Rickettsia bellii identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (1109 bp). (c) Phylogenetic tree of Rickettsia raoultii identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (855 bp). (d) Phylogenetic tree of Anaplasma identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (1455 bp) and triangles (547 bp). (e) Phylogenetic tree of Coxiella identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (1463 bp). |
Phylogenetic analysis of Rickettsia bellii
Rickettsia bellii (OP286852) was found only in I. persulcatus, and the sequence of R. bellii was randomly selected to construct the evolutionary tree. The sequence of R. bellii obtained in this study is displayed on the phylogenetic tree with squares (Fig. 9b). The phylogenetic tree of the 16S rRNA gene shows that R. bellii found in Inner Mongolia is on the same branch as R. bellii strain RML369-C (NR074484). The sequences were compared and analyzed with MEGA 7.0 software and showed that the 16S rRNA gene sequence of R. bellii from Inner Mongolia showed 94.23% homology with the R. bellii strain RML369-C (NR074484) isolated in Marseille, France.
Phylogenetic analysis of Rickettsia raoultii
The R. raoultii (OP286857) sequences were randomly selected to build a phylogenetic tree, named after regions and displayed on the phylogenetic tree with squares (Fig. 9c). The phylogenetic tree of the 16S rRNA gene shows that R. raoultii found in Inner Mongolia is on the same branch as R. raoultii strain IM16 (KY474575) and R. raoultii isolate Binxian-91 (MN446747). The sequences were compared and analyzed with MEGA 7.0 software and show that the 16S rRNA gene sequence of R. raoultii in Inner Mongolia is 100% homologous with the R. raoultii IM16 strain (KY474575) in China and R. raoultii (MN446747) found in Haemaphysalis longicornis in Bin County, Shanxi Province.
Phylogenetic analysis of Anaplasma
The Anaplasma (OP286854 and OP286856) sequences were randomly selected to build a phylogenetic tree, named after regions and displayed on the phylogenetic tree with squares and triangles (Fig. 9d). The phylogenetic tree of the 16S rRNA gene shows that Anaplasma found in Inner Mongolia is on the same branch as Anaplasma ovis strain Ao89 (KJ639879). The sequences were compared and analyzed using MEGA 7.0 software. They show that the 16S rRNA gene sequence of Anaplasma in Inner Mongolia has 99.93% homology with A. ovis strain Ao89 (KJ639879) found in the Qilian Mountains, Lanzhou.
Phylogenetic analysis of Coxiella
The Coxiella (OP286858) sequences were randomly selected to build a phylogenetic tree, named after regions and displayed on the phylogenetic tree with squares (Fig. 9e). The phylogenetic tree of the 16S rRNA gene shows that Coxiella found in Inner Mongolia is on the same branch as uncultured Coxiella sp. clones DX-56 (JX432012) and DX-68 (JX432013). The sequences were compared and analyzed using MEGA 7.0 software. The results show that the homologies between the 16S rRNA gene sequence of Coxiella in Inner Mongolia and the sequences of Coxiella DX-56 (JX432012) and Coxiella DX-68 (JX432013) are 99.93% and 99.66%, respectively.
Discussion
Dermacentor nuttalli is widely distributed in northern China. This tick is parasitic on livestock and causes severe disease in humans [24, 60]. The species carries different pathogens, such as Babesia, A. ovis, Rickettsia, and Coxiella [41, 57, 67]. Ixodes persulcatus is the dominant tick species in northeast China. Because of the large east–west span of Inner Mongolia, I. persulcatus populations exist at the border with northeast China [33]. The species is usually associated with eight species of viruses, including Alongshan virus and tick-borne encephalitis virus, which pose a serious threat to human health and safety [61].
Dermacentor nuttalli is the dominant tick species in Inner Mongolia, with a major influence on the economy and health of the local human population. Jiao et al. [18] carried out a simple microbial diversity analysis of ticks on cattle in the Hulun Buir area of Inner Mongolia. Beyond this, the microbial community composition of ticks in other areas of Inner Mongolia was not further investigated. The bacterial diversity of different tick species must be further analyzed to better understand the relationships between ticks and microorganisms. Samples collected from multiple regions are more likely to harbor new pathogens in their microbial diversity.
In our investigation, we applied PacBio full-length 16S rRNA third-generation sequencing to the V1–V9 regions of the 16S rRNA. In their study on oral microorganisms, Zhang et al. [69] found that OTU sequences generated by PacBio were much larger than those generated on the MiSeq platform. One of the advantages of third-generation sequencing is that it enables direct reading of DNA molecules without the need for PCR amplification or library construction. By avoiding these steps, third-generation sequencing can provide more realistic, comprehensive, and high-quality genome sequences [58]. Therefore, we adopted PacBio full-length 16S rRNA third-generation sequencing for the microbial diversity analysis of ticks in Inner Mongolia. According to our analysis, the microbial diversity in D. nuttalli and I. persulcatus samples from different regions of Inner Mongolia differed. The highest microbial diversity was discovered in D. nuttalli samples from Hulun Buir, where the main constituent microorganisms were Rickettsia (35.92%) and Arsenophonus (41.51%). Rickettsia is an arthropod-associated obligate intracellular gram-negative bacterium that can cause mild to severe disease in humans [10, 47]. Arsenophonus is an intracellular symbiotic bacterium of insects with a wide host range and rich biodiversity [35]. The “son killer” of the parasitic wasp Nasonia vitripennis is due to the male-killing phenotype [40]. An effect on ticks has not yet been reported, and its other biological functions have not yet been identified [29]. Studies have shown that the microbiological makeup of ticks can be influenced by a variety of factors, including tick species, life stage, sex, host blood feeding, and geographic location [23]. It is possible that the geographic location and the small number of I. persulcatus samples used in this study contributed to the increased microbial diversity discovered in D. nuttalli compared with I. persulcatus. In China, 28 species of human pathogens have been detected in I. persulcatus [61]. Using PacBio full-length 16S rRNA third-generation sequencing, only the microbial composition of bacteria in ticks was investigated in this study. Although third-generation sequencing technology has excellent coverage and read length, due to its relatively high single-molecule error rate, deep sequencing is required to obtain high-quality data results. Second-generation sequencing technologies can provide an additional level of validation, as they have higher precision and accuracy than third-generation sequencing and can detect low-frequency variants. PCR-based methods can also be used to validate the results and provide an additional level of confidence. Combining the results of multiple methods can help to improve the accuracy and reliability of pathogen identification.
Rickettsia raoultii is a pathogen of the spotted fever group, which is transmitted vertically in arthropods as a symbiotic bacterium and in vertebrates as a pathogenic bacterium of human diseases [11]. Anaplasma is a gram-negative intracellular obligate parasite, and its pathogenicity poses an important threat to several animal species and to public health [4]. Currently, there are six species of Anaplasma recognized worldwide, i.e., A. phagocytophilum, A. ovis, A. capra, A. bovis, A. marginale, and A. platys [20]. In addition to A. phagocytophilum, A. bovis and A. capra have been reported to infect humans [6, 28]. A novel typhus group Rickettsia species, Rickettsiales bacterium Ac37b, was found in the Ordos and Chifeng region [54]. The Rickettsia typhus group is composed of R. prowazekii and R. typhi. In Australia, three types of typhus, epidemic typhus, murine typhus, and tsutsugamushi disease (scrub typhus), have been found successively. These are closely related to native wild animals and ticks in Australia [13]. In China, scrub typhus has also been an important cause of human morbidity and mortality in the past decade. The disease was initially identified only in southern China, but now cases of typhus have been reported in northern China, with a wide geographical distribution [31]. Notably, Candidatus Lariskella was found in I. persulcatus in Arshan. Candidatus Lariskella was initially proposed in 2012 [36]. “Candidatus Lariskella arthropodarum” was found in samples taken from individuals in Russia who had acute febrile illnesses as well as in locally obtained I. persulcatus [38], showing its potential pathogenicity.
Through the construction of phylogenetic trees using 16S rRNA genes of the major bacteria R. bellii and main pathogenic bacteria Rickettsiales bacterium Ac37b, Anaplasma, Coxiella, and R. raoultii from this study, it was found that R. bellii showed a 94.23% homology with the R. bellii strain RML369-C (NR074484) isolated from France, Rickettsiales bacterium Ac37b had 100% homology with Rickettsiales bacterium Ac37b (CP009217) isolated from Brazil, Anaplasma had high homology with A. ovis strain Ao89 (KJ639879) in the Qilian Mountains (99.93%), Coxiella was highly homologous with Coxiella DX-56 (JX432012) (99.93%), and R. raoultii had 100% homology with R. raoultii strain IM16 (KY474575). The Qilian Mountains and Hebei are both in northern China, and Inner Mongolia is close to them. Therefore, the genetic evolution of tick-borne pathogens may be similar. Among them, both symbiotic bacteria and pathogens of the Coxiella genus can cause disease and lead to the death of humans and animals, while Coxiella burnetii is the pathogen causing Q fever [9]. However, at present, it is not clear whether ticks are of great significance in the natural transmission of C. burnetii [5]. Rickettsia bellii is the only known species in a third group that differentiated before the spotted fever group and the typhus group separated. Rickettsia bellii is the most prevalent Rickettsia species in American ticks, and it has been detected in a wide range of tick species, exhibiting the broadest arthropod host range among all known Rickettsia [26, 27, 44]. It causes slight inflammatory reactions in mammals [42]. However, the pathogenic potential for humans is still unknown and should be closely monitored [1].
It is necessary to further explore the relationship between R. bellii and other Rickettsia. In the Ordos area, pathogenic bacteria that are more threatening to humans were found, and a new classification of Rickettsia emerged. Pathogen prevention and control in this area needs to focus on monitoring and strengthening the popular knowledge of methods for personal protection. The newly discovered pathogens in Inner Mongolia need to be isolated and sequenced in future studies, and the pathogenicity of the organisms should be tested through subsequent animal experiments.
As essential vectors for carrying and spreading infections, ticks have a significant impact on the health of humans and other terrestrial vertebrates. Microbiota vaccines are used against endosymbiotic and/or nonendosymbiotic bacteria found in ticks and can help to stop the spread of ticks and/or tick-borne illnesses [63]. The findings of this study offer fundamental information for the development of a tick microbiota vaccine to stop the spread of infections carried by ticks in the Inner Mongolia Autonomous Region.
In this study, we analyzed the microbial diversity of two ticks in four regions of Inner Mongolia. A novel Rickettsia species, Rickettsiales bacterium Ac37b, was found in Inner Mongolia for the first time, and R. bellii was found in I. persulcatus. Ticks carried more potential pathogens in the Ordos area, and there were coinfections of Rickettsia and Anaplasma, which may be related to the geographical environment. Although the number of ticks collected was limited, we unveiled the microbial diversity in ticks in Inner Mongolia using third-generation sequencing. The findings of our study indicate that certain pathogens identified in ticks have the potential to cause severe human diseases. These results have major implications for researchers investigating tick-borne illnesses and can aid in the development of preventive and therapeutic strategies.
Acknowledgments
This work received financial support from the Scientific Research Project of the Mongolian Medicine Collaborative Innovation Center of the Inner Mongolia Autonomous Region (Grant MYYXTPY202206); Zhiyuan talent project of Inner Mongolia Medical University (Grant ZY0201027); Health Science and Technology Plan of Inner Mongolia Autonomous Region in 2022 (Grant 202201213); Natural Science Foundation of Inner Mongolia Autonomous Region (Grant 2022LHMS08004). We are very grateful to the Inner Mongolia Center for Disease Control and Prevention for providing tick samples. We are very grateful to the Molecular Biology Research Center of Inner Mongolia Medical University for providing experimental facilities and conditions. The authors declare that they have no conflict of interest.
References
- Abreu DPB, Peixoto MP, Luz HR, Zeringóta V, Santolin ÍDAC, Famadas KM, Faccini JLH, McIntosh D. 2019. Two for the price of one: Co-infection with Rickettsia bellii and spotted fever group Rickettsia in Amblyomma (Acari: Ixodidae) ticks recovered from wild birds in Brazil. Ticks and Tick-Borne Diseases, 10(6), 101266. [CrossRef] [PubMed] [Google Scholar]
- Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, Gaskins HR, Stumpf RM, Yildirim S, Torralba M, Gillis M, Wilson BA, Nelson KE, White BA, Leigh SR. 2013. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME Journal, 7(7), 1344–1353. [CrossRef] [PubMed] [Google Scholar]
- Belkahia H, Selmi R, Zamiti S, Daaloul-Jedidi M, Messadi L, Ben Said M. 2021. Zoonotic Rickettsia species in small ruminant ticks from Tunisia. Frontiers in Veterinary Science, 8, 676896. [CrossRef] [PubMed] [Google Scholar]
- Ben Said M, Belkahia H, Messadi L. 2018. Anaplasma spp. in North Africa: A review on molecular epidemiology, associated risk factors and genetic characteristics. Ticks and Tick-Borne Diseases, 9(3), 543–555. [CrossRef] [PubMed] [Google Scholar]
- Brenner AE, Muñoz-Leal S, Sachan M, Labruna MB, Raghavan R. 2021. Coxiella burnetii and related tick endosymbionts evolved from pathogenic ancestors. Genome Biology and Evolution, 13(7), evab108. [CrossRef] [PubMed] [Google Scholar]
- Chochlakis D, Ioannou I, Tselentis Y, Psaroulaki A. 2010. Human anaplasmosis and Anaplasma ovis variant. Emerging Infectious Diseases, 16(6), 1031–1032. [CrossRef] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Research, 37(Database issue), D141–D145. [CrossRef] [PubMed] [Google Scholar]
- Colwell DD, Dantas-Torres F, Otranto D. 2011. Vector-borne parasitic zoonoses: emerging scenarios and new perspectives. Veterinary Parasitology, 182(1), 14–21. [CrossRef] [PubMed] [Google Scholar]
- Duron O, Sidi-Boumedine K, Rousset E, Moutailler S, Jourdain E. 2015. The importance of ticks in Q fever transmission: What has (and has not) been demonstrated? Trends in Parasitology, 31(11), 536–552. [CrossRef] [PubMed] [Google Scholar]
- El Karkouri K, Ghigo E, Raoult D, Fournier PE. 2022. Genomic evolution and adaptation of arthropod-associated Rickettsia. Scientific Reports, 12(1), 3807. [CrossRef] [PubMed] [Google Scholar]
- El Karkouri K, Kowalczewska M, Armstrong N, Azza S, Fournier PE, Raoult D. 2017. Multi-omics analysis sheds light on the evolution and the intracellular lifestyle strategies of spotted fever group Rickettsia spp. Frontiers in Microbiology, 8, 1363. [CrossRef] [PubMed] [Google Scholar]
- Fang LQ, Liu K, Li XL, Liang S, Yang Y, Yao HW, Sun RX, Sun Y, Chen WJ, Zuo SQ, Ma MJ, Li H, Jiang JF, Liu W, Yang XF, Gray GC, Krause PJ, Cao WC. 2015. Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infectious diseases, 15(12), 1467–1479. [CrossRef] [Google Scholar]
- Graves S, Stenos J. 2009. Rickettsioses in Australia. Annals of the New York Academy of Sciences, 1166, 151–155. [CrossRef] [PubMed] [Google Scholar]
- Guo WP, Huang B, Zhao Q, Xu G, Liu B, Wang YH, Zhou EM. 2018. Human-pathogenic Anaplasma spp., and Rickettsia spp. in animals in Xi’an, China. PLoS Neglected Tropical Diseases, 12(11), e0006916. [CrossRef] [PubMed] [Google Scholar]
- Hoskins JD. 1991. Ixodid and argasid ticks. Keys to their identification. Veterinary Clinics of North America. Small Animal Practice, 21(1), 185–197. [CrossRef] [Google Scholar]
- Ijaz MU, Ahmed MI, Zou X, Hussain M, Zhang M, Zhao F, Xu X, Zhou G, Li C. 2018. Beef, casein, and soy proteins differentially affect lipid metabolism, triglycerides accumulation and gut microbiota of high-fat diet-fed C57BL/6J mice. Frontiers in Microbiology, 9, 2200. [CrossRef] [PubMed] [Google Scholar]
- Jia N, Zheng YC, Ma L, Huo QB, Ni XB, Jiang BG, Chu YL, Jiang RR, Jiang JF, Cao WC. 2014. Human infections with Rickettsia raoultii, China. Emerging Infectious Diseases, 20(5), 866–868. [CrossRef] [PubMed] [Google Scholar]
- Jiao J, Lu Z, Yu Y, Ou Y, Fu M, Zhao Y, Wu N, Zhao M, Liu Y, Sun Y, Wen B, Zhou D, Yuan Q, Xiong X. 2021. Identification of tick-borne pathogens by metagenomic next-generation sequencing in Dermacentor nuttalli and Ixodes persulcatus in Inner Mongolia, China. Parasites & Vectors, 14(1), 287. [CrossRef] [PubMed] [Google Scholar]
- Jongejan F, Uilenberg G. 2004. The global importance of ticks. Parasitology, 129(Suppl), S3–S14. [CrossRef] [PubMed] [Google Scholar]
- Kocan KM, de la Fuente J, Cabezas-Cruz A. 2015. The genus Anaplasma: new challenges after reclassification. Revue Scientifique et Technique (International Office of Epizootics), 34(2), 577–586. [PubMed] [Google Scholar]
- Körner S, Makert GR, Ulbert S, Pfeffer M, Mertens-Scholz K. 2021. The prevalence of Coxiella burnetii in hard ticks in Europe and their role in Q Fever transmission revisited – A Systematic review. Frontiers in Veterinary Science, 8, 655715. [CrossRef] [PubMed] [Google Scholar]
- Köseoğlu AE, Can H, Güvendi M, Erkunt Alak S, Kandemir Ç, Taşkın T, Demir S, Akgül G, Değirmenci Döşkaya A, Karakavuk M, Döşkaya M, Gürüz AY, Ün C. 2021. Molecular investigation of bacterial and protozoal pathogens in ticks collected from different hosts in Turkey. Parasites & vectors, 14(1), 270. [CrossRef] [PubMed] [Google Scholar]
- Kueneman JG, Esser HJ, Weiss SJ, Jansen PA, Foley JE. 2021. Tick microbiomes in neotropical forest fragments are best explained by tick-associated and environmental factors rather than host blood source. Applied and Environmental Microbiology, 87(7), e02668-20. [CrossRef] [PubMed] [Google Scholar]
- Kulakova NV, Khasnatinov MA, Sidorova EA, Adel’ Shin RV, Belikov SI. 2014. Molecular identification and phylogeny of Dermacentor nuttalli (Acari: Ixodidae). Parasitology Research, 113(5), 1787–1793. [CrossRef] [PubMed] [Google Scholar]
- Kumar D, Downs LP, Adegoke A, Machtinger E, Oggenfuss K, Ostfeld RS, Embers M, Karim S. 2022. An exploratory study on the microbiome of Northern and Southern populations of Ixodes scapularis ticks predicts changes and unique bacterial interactions. Pathogens, 11(2), 130. [CrossRef] [PubMed] [Google Scholar]
- Labruna MB, McBride JW, Bouyer DH, Camargo LM, Camargo EP, Walker DH. 2004. Molecular evidence for a spotted fever group Rickettsia species in the tick Amblyomma longirostre in Brazil. Journal of Medical Entomology, 41(3), 533–537. [CrossRef] [PubMed] [Google Scholar]
- Labruna MB, Whitworth T, Bouyer DH, McBride J, Camargo LM, Camargo EP, Popov V, Walker DH. 2004. Rickettsia bellii and Rickettsia amblyommii in Amblyomma ticks from the State of Rondônia, Western Amazon, Brazil. Journal of Medical Entomology, 41(6), 1073–1081. [CrossRef] [PubMed] [Google Scholar]
- Li H, Zheng YC, Ma L, Jia N, Jiang BG, Jiang RR, Huo QB, Wang YW, Liu HB, Chu YL, Song YD, Yao NN, Sun T, Zeng FY, Dumler JS, Jiang JF, Cao WC. 2015. Human infection with a novel tick-borne Anaplasma species in China: a surveillance study. Lancet Infectious Diseases, 15(6), 663–670. [CrossRef] [Google Scholar]
- Lo WS, Huang YY, Kuo CH. 2016. Winding paths to simplicity: genome evolution in facultative insect symbionts. FEMS Microbiology Reviews, 40(6), 855–874. [CrossRef] [PubMed] [Google Scholar]
- Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. 2011. UniFrac: an effective distance metric for microbial community comparison. ISME Journal, 5(2), 169–172. [CrossRef] [PubMed] [Google Scholar]
- Lu M, Li F, Liao Y, Shen JJ, Xu JM, Chen YZ, Li JH, Holmes EC, Zhang YZ. 2019. Epidemiology and diversity of rickettsiales bacteria in humans and animals in Jiangsu and Jiangxi provinces, China. Scientific Reports, 9(1), 13176. [CrossRef] [PubMed] [Google Scholar]
- Luan Y, Gou J, Zhong D, Ma L, Yin C, Shu M, Liu G, Lin Q. 2023. The tick-borne pathogens: An overview of China’s situation. Acta Parasitologica, 68(1), 1–20. [CrossRef] [PubMed] [Google Scholar]
- Ma B, Ma XY, Zhang Y, Chen HB, Wang Q, Li LH. 2021. Prediction of suitable habitats of Ixodes persulcatus in China. Chinese Journal of Schistosomiasis Control, 33(2), 169–176 [in Chinese]. [Google Scholar]
- Maltezou HC, Andonova L, Andraghetti R, Bouloy M, Ergonul O, Jongejan F, Kalvatchev N, Nichol S, Niedrig M, Platonov A, Thomson G, Leitmeyer K, Zeller H. 2010. Crimean-Congo hemorrhagic fever in Europe: current situation calls for preparedness. Eurosurveillance, 15(10), 19504. [CrossRef] [Google Scholar]
- Massey JH, Newton ILG. 2022. Diversity and function of arthropod endosymbiont toxins. Trends in Microbiology, 30(2), 185–198. [CrossRef] [PubMed] [Google Scholar]
- Matsuura Y, Kikuchi Y, Meng XY, Koga R, Fukatsu T. 2012. Novel clade of alphaproteobacterial endosymbionts associated with stinkbugs and other arthropods. Applied and Environmental Microbiology, 78(12), 4149–4156. [CrossRef] [PubMed] [Google Scholar]
- Mead P, Hinckley A, Hook S, Beard CB. 2015. TickNET-A collaborative public health approach to tickborne disease surveillance and research. Emerging Infectious Diseases, 21(9), 1574–1577. [CrossRef] [PubMed] [Google Scholar]
- Mediannikov O, Ivanov LI, Nishikawa M, Saito R, Sidel’nikov IuN, Zdanovskaia NI, Mokretsova EV, Tarasevich IV, Suzuki H. 2004. Microorganism “Montezuma” of the order Rickettsiales: the potential causative agent of tick-borne disease in the Far East of Russia. Zhurnal Mikrobiologii, Epidemiologii i Immunobiologii, 1, 7–13. [Google Scholar]
- Moreno C, Romero J, Espejo RT. 2002. Polymorphism in repeated 16S rRNA genes is a common property of type strains and environmental isolates of the genus vibrio. Microbiology, 148(Pt 4), 1233–1239. [CrossRef] [PubMed] [Google Scholar]
- Nadal-Jimenez P, Griffin JS, Davies L, Frost CL, Marcello M, Hurst GDD. 2019. Genetic manipulation allows in vivo tracking of the life cycle of the son-killer symbiont, Arsenophonus nasoniae, and reveals patterns of host invasion, tropism and pathology. Environmental Microbiology, 21(8), 3172–3182. [CrossRef] [PubMed] [Google Scholar]
- Ni J, Lin H, Xu X, Ren Q, Aizezi M, Luo J, Luo Y, Ma Z, Chen Z, Tan Y, Guo J, Liu W, Qu Z, Wu Z, Wang J, Li Y, Guan G, Luo J, Yin H, Liu G. 2020. Coxiella burnetii is widespread in ticks (Ixodidae) in the Xinjiang areas of China. BMC Veterinary Research, 16(1), 317. [CrossRef] [PubMed] [Google Scholar]
- Ogata H, La Scola B, Audic S, Renesto P, Blanc G, Robert C, Fournier PE, Claverie JM, Raoult D. 2006. Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. Plos Genetics, 2(5), e76. [CrossRef] [PubMed] [Google Scholar]
- Parola P, Paddock CD, Socolovschi C, Labruna MB, Mediannikov O, Kernif T, Abdad MY, Stenos J, Bitam I, Fournier PE, Raoult D. 2013. Update on tick-borne rickettsioses around the world: a geographic approach. Clinical Microbiology Reviews, 26(4), 657–702. [CrossRef] [PubMed] [Google Scholar]
- Philip RN, Casper EA. 1981. Serotypes of spotted fever group rickettsiae isolated from Dermacentor andersoni (Stiles) ticks in western Montana. American Journal of Tropical Medicine and Hygiene, 30(1), 230–238. [CrossRef] [PubMed] [Google Scholar]
- Poirier S, Rué O, Peguilhan R, Coeuret G, Zagorec M, Champomier-Vergès MC, Loux V, Chaillou S. 2018. Deciphering intra-species bacterial diversity of meat and seafood spoilage microbiota using gyrB amplicon sequencing: A comparative analysis with 16S rDNA V3–V4 amplicon sequencing. PLoS One, 13(9), e0204629. [CrossRef] [PubMed] [Google Scholar]
- Randolph SE. 2008. Tick-borne encephalitis incidence in Central and Eastern Europe: consequences of political transition. Microbes and Infection, 10(3), 209–216. [CrossRef] [PubMed] [Google Scholar]
- Raoult D, Roux V. 1997. Rickettsioses as paradigms of new or emerging infectious diseases. Clinical Microbiology Reviews, 10(4), 694–719. [CrossRef] [PubMed] [Google Scholar]
- Rosenberg R, Lindsey NP, Fischer M, Gregory CJ, Hinckley AF, Mead PS, Paz-Bailey G, Waterman SH, Drexler NA, Kersh GJ, Hooks H, Partridge SK, Visser SN, Beard CB, Petersen LR. 2018. Vital signs: Trends in reported vectorborne disease cases – United States and territories, 2004–2016. Morbidity and Mortality Weekly Report, 67(17), 496–501. [CrossRef] [PubMed] [Google Scholar]
- Ruiling Z, Zhendong H, Guangfu Y, Zhong Z. 2019. Characterization of the bacterial community in Haemaphysalis longicornis (Acari: Ixodidae) throughout developmental stages. Experimental and Applied Acarology, 77(2), 173–186. [CrossRef] [PubMed] [Google Scholar]
- Sasaki K, Honma M, Nakao M, Sasaki M, Hashimoto Y, Ishida-Yamamoto A, Yoshii K. 2021. Survey to detect tick-borne encephalitis virus from human-feeding ticks in Hokkaido, Japan. Journal of Dermatology, 48(7), 1094–1097. [CrossRef] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23), 7537–7541. [CrossRef] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biology, 12(6), R60. [CrossRef] [PubMed] [Google Scholar]
- Senbill H, Tanaka T, Karawia D, Rahman S, Zeb J, Sparagano O, Baruah A. 2022. Morphological identification and molecular characterization of economically important ticks (Acari: Ixodidae) from North and North-Western Egypt. Acta Tropica, 231, 106438. [CrossRef] [PubMed] [Google Scholar]
- Shpynov SN, Fournier PE, Pozdnichenko NN, Gumenuk AS, Skiba AA. 2018. New approaches in the systematics of rickettsiae. New Microbes and New Infections, 23, 93–102. [CrossRef] [PubMed] [Google Scholar]
- Simon JA, Marrotte RR, Desrosiers N, Fiset J, Gaitan J, Gonzalez A, Koffi JK, Lapointe FJ, Leighton PA, Lindsay LR, Logan T, Milord F, Ogden NH, Rogic A, Roy-Dufresne E, Suter D, Tessier N, Millien V. 2014. Climate change and habitat fragmentation drive the occurrence of Borrelia burgdorferi, the agent of Lyme disease, at the northeastern limit of its distribution. Evolutionary Applications, 7(7), 750–764. [CrossRef] [PubMed] [Google Scholar]
- Singer E, Bushnell B, Coleman-Derr D, Bowman B, Bowers RM, Levy A, Gies EA, Cheng JF, Copeland A, Klenk HP, Hallam SJ, Hugenholtz P, Tringe SG, Woyke T. 2016. High-resolution phylogenetic microbial community profiling. ISME Journal, 10(8), 2020–2032. [CrossRef] [PubMed] [Google Scholar]
- Song R, Wang Q, Guo F, Liu X, Song S, Chen C, Tu C, Wureli H, Wang Y. 2018. Detection of Babesia spp., Theileria spp. and Anaplasma ovis in Border Regions, northwestern China. Transboundary and Emerging Diseases, 65(6), 1537–1544. [CrossRef] [PubMed] [Google Scholar]
- van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. 2018. The third revolution in sequencing technology. Trends in Genetics, 34(9), 666–681. [CrossRef] [PubMed] [Google Scholar]
- Walker JB. 1991. A review of the ixodid ticks (Acari, Ixodidae) occurring in southern Africa. Onderstepoort Journal of Veterinary Research, 58(2), 81–105. [Google Scholar]
- Wang F, Wang D, Guo G, Hu Y, Wei J, Liu J. 2019. Species delimitation of the Dermacentor ticks based on phylogenetic clustering and niche modeling. PeerJ, 7, e6911. [CrossRef] [PubMed] [Google Scholar]
- Wang SS, Liu JY, Wang BY, Wang WJ, Cui XM, Jiang JF, Sun Y, Guo WB, Pan YS, Zhou YH, Lin ZT, Jiang BG, Zhao L, Cao WC. 2023. Geographical distribution of Ixodes persulcatus and associated pathogens: Analysis of integrated data from a China field survey and global published data. One Health, 16, 100508. [CrossRef] [PubMed] [Google Scholar]
- Wang YZ, Mu LM, Zhang K, Yang MH, Zhang L, Du JY, Liu ZQ, Li YX, Lu WH, Chen CF, Wang Y, Chen RG, Xu J, Yuan L, Zhang WJ, Zuo WZ, Shao RF. 2015. A broad-range survey of ticks from livestock in Northern Xinjiang: changes in tick distribution and the isolation of Borrelia burgdorferi sensu stricto. Parasites & Vectors, 8, 449. [CrossRef] [PubMed] [Google Scholar]
- Wu-Chuang A, Hodžić A, Mateos-Hernández L, Estrada-Peña A, Obregon D, Cabezas-Cruz A. 2021. Current debates and advances in tick microbiome research. Current Research in Parasitology & Vector-Borne Diseases, 1, 100036. [CrossRef] [PubMed] [Google Scholar]
- Wu XB, Na RH, Wei SS, Zhu JS, Peng HJ. 2013. Distribution of tick-borne diseases in China. Parasites & Vectors, 6, 119. [CrossRef] [PubMed] [Google Scholar]
- Yamaji K, Aonuma H, Kanuka H. 2018. Distribution of tick-borne diseases in Japan: Past patterns and implications for the future. Journal of Infection and Chemotherapy, 24(7), 499–504. [CrossRef] [PubMed] [Google Scholar]
- Yang J, Liu Z, Guan G, Che R, Niu Q, Li Y, Liu J, Ma M, Ren Q, Liu A, Luo J, Yin H. 2012. Evaluation of molecular methods for detection of Borrelia burgdorferi senso lato in ticks. Diagnostic Microbiology and Infectious Disease, 73(1), 80–83. [CrossRef] [PubMed] [Google Scholar]
- Yin X, Guo S, Ding C, Cao M, Kawabata H, Sato K, Ando S, Fujita H, Kawamori F, Su H, Shimada M, Shimamura Y, Masuda S, Ohashi N. 2018. Spotted Fever Group Rickettsiae in Inner Mongolia, China, 2015–2016. Emerging Infectious Diseases, 24(11), 2105–2107. [CrossRef] [PubMed] [Google Scholar]
- Yu Z, Wang H, Wang T, Sun W, Yang X, Liu J. 2015. Tick-borne pathogens and the vector potential of ticks in China. Parasites & Vectors, 8, 24. [CrossRef] [PubMed] [Google Scholar]
- Zhang J, Su L, Wang Y, Deng S. 2020. Improved high-throughput sequencing of the human oral microbiome: From Illumina to PacBio. Canadian Journal of Infectious Diseases & Medical Microbiology, 2020, 6678872. [Google Scholar]
- Zhang YK, Zhang XY, Liu JZ. 2019. Ticks (Acari: Ixodoidea) in China: Geographical distribution, host diversity, and specificity. Archives of Insect Biochemistry and Physiology, 102(3), e21544. [CrossRef] [PubMed] [Google Scholar]
- Zhao GP, Wang YX, Fan ZW, Ji Y, Liu MJ, Zhang WH, Li XL, Zhou SX, Li H, Liang S, Liu W, Yang Y, Fang LQ. 2021. Mapping ticks and tick-borne pathogens in China. Nature Communications, 12(1), 1075. [CrossRef] [PubMed] [Google Scholar]
- Zhao JW, Wang HY, Wang Y. 2012. Regional distribution profiles of tick-borne pathogens in China. Chinese Journal of Vector Biology and Control, 23(5), 445–448. [Google Scholar]
Cite this article as: Su S, Hong M, Cui M-Y, Gui Z, Ma S-F, Wu L, Xing L-L, Mu L, Yu J-F, Fu S-Y, Gao R-J & Qi D-D. 2023. Microbial diversity of ticks and a novel typhus group Rickettsia species (Rickettsiales bacterium Ac37b) in Inner Mongolia, China. Parasite 30, 58.
All Tables
All Figures
|
Figure 1 Map of tick collection sites in Inner Mongolia, China. |
| In the text | |
|
Figure 2 Shannon–Wiener curve. X-axis: amount of sequencing data; Y-axis: corresponding Shannon diversity index. |
| In the text | |
|
Figure 3 Microbe composition of different regions and species. (a) Top 10 microbial components at the genus level. (b) Top 12 microbial components at the species level. |
| In the text | |
|
Figure 4 Alpha diversity measures for Dermacentor nuttalli and Ixodes persulcatus in four areas. (a) Shannon’s index. (b) Simpson’s index. |
| In the text | |
|
Figure 5 (a) Clustering tree analysis by linear discriminant analysis effect size (LEfSe). (b) Histogram of LDA analysis. |
| In the text | |
|
Figure 6 PCoA of β-diversity measures for twelve groups. Weighted UniFrac PCoA graph showing PC1, which accounts for 47.46% of variation, and PC2, which accounts for 28.93% of variation. Different colored dots represent different regions and species. |
| In the text | |
|
Figure 7 Venn diagram for cluster analysis of OTUs between Dermacentor nuttalli and Ixodes persulcatus (genus level). |
| In the text | |
|
Figure 8 The composition of bacteria and pathogenic bacteria with a high abundance. |
| In the text | |
|
Figure 9 Phylogenetic tree of Rickettsiales bacterium Ac37b, Rickettsia bellii, Rickettsia raoultii, Anaplasma and Coxiella in ticks based on neighbor-joining (NJ) modeling; only values higher than 60 were added to the tree branches. (a) Phylogenetic tree of Rickettsiales bacterium Ac37b identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (1320 bp) and triangles (1430 bp). (b) Phylogenetic tree of Rickettsia bellii identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (1109 bp). (c) Phylogenetic tree of Rickettsia raoultii identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (855 bp). (d) Phylogenetic tree of Anaplasma identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (1455 bp) and triangles (547 bp). (e) Phylogenetic tree of Coxiella identified in Inner Mongolia; the 16S rRNA gene sequences obtained in this study are marked with black squares (1463 bp). |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.