| Issue |
Parasite
Volume 22, 2015
|
|
|---|---|---|
| Article Number | 39 | |
| Number of page(s) | 6 | |
| DOI | https://doi.org/10.1051/parasite/2015039 | |
| Published online | 21 December 2015 | |
Research Article
Interferon gamma effect on immune mediator production in human nerve cells infected by two strains of Toxoplasma gondii
Effet de l’interféron gamma sur la production des médiateurs immuns par les cellules nerveuses humaines infectées par deux souches de Toxoplasma gondii
1
Univ. Limoges, UMR-S 1094, Tropical Neuroepidemiology, Institute of Neuroepidemiology and Tropical Neurology, CNRS FR 3503 GEIST, 87000
Limoges, France
2
UMR CNRS 7276, FR 3503 GEIST, Faculty of Pharmacy, University of Limoges, 87000
Limoges, France
3
CHU Limoges, Department of Parasitology, and Biological Resource Centre for Toxoplasma, 87000
Limoges, France
* Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
7
October
2015
Accepted:
29
November
2015
Abstract
Interferon gamma (IFN-γ) is the major immune mediator that prevents toxoplasmic encephalitis in murine models. The lack of IFN-γ secretion causes reactivation of latent T. gondii infection that may confer a risk for severe toxoplasmic encephalitis. We analyse the effect of IFN-γ on immune mediator production and parasite multiplication in human nerve cells infected by tachyzoites of two T. gondii strains (RH and PRU). IFN-γ decreased the synthesis of MCP-1, G-CSF, GM-CSF and Serpin E1 in all cell types. It decreased IL-6, migration inhibitory factor (MIF) and GROα synthesis only in endothelial cells, while it increased sICAM and Serpin E1 synthesis only in neurons. The PRU strain burden increased in all nerve cells and in contrast, RH strain replication was controlled in IFN-γ-stimulated microglial and endothelial cells but not in IFN-γ-stimulated neurons. The proliferation of the PRU strain in all stimulated cells could be a specific effect of this strain on the host cell.
Résumé
L’interféron gamma (IFN-γ) est le médiateur immunitaire majeur qui empêche le développement de l’encéphalite toxoplasmique dans le modèle murin. L’absence de sécrétion d’IFN-γ provoque la réactivation de l’infection latente à T. gondii, avec un risque de développement d’une toxoplasmose cérébrale sévère. Nous avons analysé l’effet de l’IFN-γ sur la production des médiateurs immunitaires et la multiplication parasitaire dans des cellules nerveuses humaines infectées par des tachyzoïtes de deux souches de T. gondii (RH et PRU). L’IFN-γ diminue la synthèse de MCP-1, G-CSF, GM-CSF et Serpin E1 dans tous les types cellulaires. Il diminue la synthèse d’IL-6, de MIF et de GROα uniquement dans les cellules endothéliales, tandis qu’il stimule la synthèse de sICAM et de Serpin E1 dans les neurones. La charge parasitaire de la souche PRU augmente dans toutes les cellules nerveuses. En revanche, la multiplication de la souche RH est contrôlée dans les cellules microgliales et endothéliales stimulées par l’IFN-γ, mais pas dans les neurones stimulés par l’IFN-γ. La prolifération de la souche PRU dans toutes les cellules stimulées pourrait être un effet spécifique de cette souche sur la cellule hôte.
Key words: Toxoplasma gondii / Human nerve cells / IFN-γ / Immunity
© N. Mammari et al., published by EDP Sciences, 2015
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Toxoplasma gondii induces a potent cellular immune response that is essential for controlling infection. Interferon gamma (IFN-γ) is one of the most important cytokines for immune control of Toxoplasma infection in mice. In infected mice, this cytokine could control tachyzoite proliferation, thus maintaining latency of chronic infection in the brain and preventing toxoplasmic encephalitis [26]. During acute Toxoplasma infection, natural killer (NK) cells, CD4+ and CD8+ T cells are the major sources of IFN-γ and this cytokine might stimulate all effector cells to activate a protective immune response against T. gondii infection [7]. In the murine central nervous system (CNS), IFN-γ activates neuronal cells to control tachyzoite multiplication [25]. The stimulation of murine microglial cells with IFN-γ and TNFα inhibited the penetration of PLK tachyzoites (mouse non-virulent type II Toxoplasma strain) [14]. Intracellular type I and II tachyzoite proliferation is inhibited by pre-treatment with IFN-γ of both human and murine astrocyte cells [15, 20]. In addition, it was demonstrated that IFN-γ is involved in Toxoplasma cyst formation in murine astrocytes and neurons [12, 18].
Most of these data were obtained in murine models. In a previous paper, we reported the production of cytokines and chemokines by infected human nerve cells in the absence of IFN-γ [19]. To further study the role of IFN-γ in the control of Toxoplasma infection in human nerve cells in vitro, we report the effect of IFN-γ on the activation of infected human nerve cells and the control of intracellular multiplication of two T. gondii strains.
Materials and methods
Parasite production
One type I strain (RH) and one type II strain (PRU) of T. gondii were used in the present study. Tachyzoites of each strain were grown and purified in human fibroblastic cell cultures (MRC5) as previously described [19]. All protocols involving animals were approved by the Committee on the Ethics of Animal Experiments of Limousin, France (Permit No. 3-07-2012).
Human nerve cell cultures
Human microglial cells (CMH5) (kindly provided by Pr. P. Vincendeau, Bordeaux, France) [16], human bone marrow endothelial cells (Hbmec) (obtained from the cell line established by Pr. D. Paulin, University of Paris 7, France) [24] and human neuroblastoma cells (SH SY5Y) (kindly provided by Pr. M.O. Jauberteau-Marchan, Limoges, France) [21] were cultured following the protocol described in the previous study [19].
Treatment and infection of human nerve cells
Each type of cell culture was treated with IFN-γ (100 ng/mL) (Sigma-Aldrich) for 24 h. A total of 106 human nerve cells (confluent cells) were then infected separately for 24 h by tachyzoites either from the RH strain (type I) or from the PRU strain (type II) with a ratio of one cell for two tachyzoites. Control cells were uninfected and non-stimulated cells, and uninfected stimulated cells. Each experiment was performed in triplicate at separate times to ensure reproducibility of results.
Cytokine, chemokine and growth factor analysis
The pro-inflammatory proteins were analysed in co-culture supernatants using the Proteome Profiler Array technique (R&D, Lille, France), following the protocol described in the previous study [19].
Quantification of parasite burden
The viable parasite burden was quantified in the pellet cell culture by semi-quantitative reverse transcription polymerase chain reaction (RT-PCR) using specific primers for T. gondii 529 gene repeat. qRT-PCR was performed using a One Step SYBR Green RT-PCR Kit (Qiagen, Paris, France). Each RNA sample (≤ 100 ng) was added to PCR tubes containing SYBR master mix (12.5 μL), RT mix (0.25 μL) and specific primers (0.6 μM) [sense: 5′-AGGCGAGGTGAGGATGA-3′; antisense: 5′-TCGTCTCGTCTGGATCGAAT-3′] (Sigma-Aldrich) [4]. Quantification was performed using a dose of tachyzoites ranging from 5 × 104 to 1 × 104 Toxoplasma amplified by primers for the 529 pb repeat gene [19].
Statistical analysis
Statistical analysis of pro-inflammatory protein profiles and comparison of parasite burden quantification were carried out using the Scheirer-Ray-Hare test. All interactions between the different factors were performed using Steel-Dwass and Siegel-Castellan tests with a 95% confidence level.
Results
Among the 36 immune mediators tested, two interleukins (IL-6 and IL-8), three chemokines (MCP-1, migration inhibitory factor [MIF], GROα) and five other markers (G-CSF, GM-CSF, Serpin E1 and sICAM) were significantly expressed in IFN-γ stimulated and non-stimulated CMH5, Hbmec and SH SY5Y infected cells [19].
Compared to immune mediator expression in infected non-stimulated cells [19], we observed that stimulation by IFN-γ leads to a decrease in IL-6 and GROα levels in RH- and PRU-infected endothelial cells (p < 0.05) (Table 1) and MCP-1, G-CSF, GM-CSF and Serpin E1 synthesis in the three cell types (Table 1).
Cell types, parasite strains and IFN-γ effects on pro-inflammatory protein expressions. Pro-inflammatory proteins were analysed in supernatants of cultivated RH- or PRU-infected cells using Proteome Profiler Array. All spots were quantified as pixel units by GeneTools. Quantification corresponds to the pixel values of uninfected cells (negative control) subtracted from pixel values of infected cells. The effect level was determined using the Scheirer-Ray-Hare test and Steel-Dwass and/or Siegel-Castellan post hoc tests. ↘ expression decrease with p < 0.05; ↘↘ expression decrease with p < 0.001. ↗ expression increase with p < 0.05; ↗↗ expression increase with p < 0.001.
More specifically, the level of IL-6 synthesis is decreased only in RH- and PRU-infected and stimulated endothelial cells (p < 0.05) (Table 1). For chemokines, we noted that MIF is decreased in stimulated and PRU-infected endothelial cells (p < 0.05) (Table 1). Monocyte chemoattractant protein 1 (MCP-1) synthesis is decreased in IFN-γ-stimulated and PRU-infected microglial cells (CMH5) (p < 0.001) and endothelial cells (Hbmec) (p < 0.001). In neurons (SH SY5Y), MCP-1 levels decreased only in RH-infected and stimulated cells (p < 0.05) (Table 1). In the presence of IFN-γ, G-CSF synthesis decreased significantly in RH- and PRU-infected microglial (p < 0.001) and endothelial cells (p < 0.001) and only in PRU-infected neurons (p < 0.05). GM-CSF production was significantly decreased in PRU-infected microglial cells (p < 0.001) and in RH-infected endothelial cells (p < 0.05) and PRU-infected neurons (p < 0.05). In the presence of IFN-γ, we also noted that sICAM, adhesion protein decreased synthesis in PRU-infected microglial (p < 0.001) and endothelial cells (p < 0.001) (Table 1) but increased in RH- and PRU-infected neurons (p < 0.001). IFN-γ also had a significant effect on Serpin E1 protein production; it decreased synthesis in PRU-infected microglial (p < 0.001) and endothelial cells (p < 0.001) and increased in PRU- and RH-infected neurons (p < 0.001). In contrast, IFN-γ did not have a significant effect on IL-8.
At 24 h post-infection, the parasite burden of the RH strain was significantly decreased in IFN-γ-stimulated endothelial cells (p < 0.01), but it increased in IFN-γ-stimulated neurons (p < 0.01). In contrast, the PRU burden rate remained high in each type of IFN-γ-stimulated cell (p < 0.01) (Fig. 1).
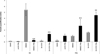 |
Figure 1. Effect of IFN-γ on parasite multiplication. Microglial cells, endothelial cells and neurons were stimulated by IFN-γ before being infected with the RH or PRU strain of T. gondii for 24 h. Parasite burden was quantified by semi-quantitative RT-PCR in stimulated and infected cells (black) and in non-stimulated and infected cells (grey). Parasite quantification was evaluated as follows: final number of tachyzoites/initial number of tachyzoites. Comparison was performed using the Scheirer-Ray-Hare test. **p < 0.01. |
Discussion
Results obtained in this study showed that stimulation by IFN-γ had a significant effect on expression of these immune mediators, decreasing their production compared to non-stimulated infected cells. This decrease was more marked in PRU-infected cells.
More specifically, in PRU-infected microglial cells, IFN-γ caused a decrease of MCP-1, growth factors (G-CSF and GM-CSF), Serpin E1 and sICAM, whereas in microglial cells infected by RH strain, IFN-γ caused a significant decrease of only G-CSF. The chemokine MCP-1 is known for initiating monocyte/macrophage activation which can promote pro-inflammatory immune response against T. gondii and it can also participate in the control of Toxoplasma infection, as described in human MRC5 fibroblasts [3] and human astrocytes [2]. The decrease of this production under IFN-γ treatment in PRU-infected cells suggests that this cytokine may control a pro-inflammatory immune reaction in the course of a type II infection. This process was confirmed by a decrease of G-CSF and GM-CSF. These two growth factors are involved in dendritic cell activation, granulocyte survival and enhancement of macrophage/microglial function so that the decrease of their production might limit immune cell activation. In addition, these two growth factors were reported to inhibit neutrophil apoptosis during Toxoplasma infection [6]. The decrease of growth factor production during PRU infection could promote neutrophil apoptosis and thus reduce the pro-inflammatory reaction induced by this strain. Serpin E1, that also decreased after IFN-γ stimulation of PRU-infected microglial cells, was reported to inhibit the uPA/uPAR pathway which promotes the secretion of MMP-9 active forms by Toxoplasma-infected macrophages [23]. After stimulation by IFN-γ, the decrease of Serpin E1 production might promote parasite invasion in these cells.
In PRU-infected endothelial cells, we noted a decrease of IL-6, and of chemokines MCP-1, MIF and GROα, of G-CSF, of Serpin E1, and sICAM. Interleukin-6 contributes to the intracellular control of type I and II parasite strains in mouse neurons [17]. The decrease of IL-6 might be involved in loss of PRU multiplication in these cells. In addition, the decrease of chemokine production could control local inflammation and promote parasite multiplication. MIF is considered as an important protein. It has been described that MIF had a role in controlling Toxoplasma parasite burdens via a strong immune inflammation response responsible for intestinal tissue damages. MIF-deficient mice were susceptible to T. gondii infection and presented a high parasite burden [13]. In our case, the high parasite burden noted in PRU-infected endothelial cells is facilitated via Serpin E1 and sICAM reduction. In RH-infected endothelial cells, IFN-γ caused a decrease of mostly the same proteins (IL-6, G-CSF and GM-CSF) except MCP-1, MIF, GROα, Serpin E1 and sICAM. These data indicate that the RH strain could cause less local pro-inflammatory reaction than the PRU strain and the normal expression of Serpin E1 and sICAM suggests a role of these proteins in the control of RH strain multiplication in endothelial cells.
In PRU-infected neurons, IFN-γ decreased only growth factors (G-CSF and GM-CSF), but increased sICAM and Serpin E1 production. A different situation was observed when neurons were infected with the RH strain: IFN-γ also increased sICAM and Serpin E1 production, but, contrary to PRU, it increased G-CSF. Moreover, a decrease in MCP-1 levels was observed in RH-infected neurons. The decrease of growth factor production during PRU infection could control pro-inflammatory reactions and promote neutrophil apoptosis of neurons infected by PRU. The high expression of Serpin E1 promotes parasite invasion and multiplication, which could be explained by the high parasite burden in PRU- and RH-infected neurons. For sICAM, this protein was known to have an inhibitory effect on T. gondii transmigration across BeWo (human placenta), Caco-2 (human intestine) and MDCK (canine kidney) cells, which can control parasite infiltration [1]. This result might indicate that sICAM could be involved in maintaining infection chronicity and Toxoplasma cyst formation [11].
In neurons stimulated by IFN-γ, both RH and PRU multiplication rates remained high at 24 h post-infection, suggesting that this cytokine alone is not sufficient for the control of parasite multiplication in neurons. The effector molecules inducing anti-toxoplasmic activity in neurons have not been specifically identified. In this study, sICAM and Serpin E1 expression indicated that they could be implicated in the increase of intra-neuronal parasite burden. Importantly, in the murine model, astrocytes help neurons to target the parasite, both cell types also serve as immunoregulators via TGF-β signalling pathways [5]. So, we can suggest that neurons alone cannot control tachyzoite multiplication. In contrast, the RH multiplication rate was decreased in human endothelial cells stimulated by IFN-γ, suggesting the implication of IFN-γ in the control of RH multiplication in these human cells. In vivo, this effect could be promoted via IFN-γ-activated lymphocytes which had a major role in parasite proliferation control. These cells can activate Th1 immune response and NK cells to limit parasite proliferation in the murine model [10].
In human microglial cells, RH-parasite burden was controlled in unstimulated and stimulated cells. It is known that IFN-γ activates microglial cells to produce NO, which can reduce and control RH intracellular multiplication in the murine model [22]. So, human microglial cells may have the capacity to control T. gondii type I strain; the addition of IFN-γ to these infected cells provokes a decrease only in growth factors, which can explain that this cytokine could control the local inflammatory reaction in these infected cells. Unexpectedly, this control of multiplication by IFN-γ in human microglial and endothelial cells was not observed for the type II strain. The PRU burden rate remained high in each type of IFN-γ-stimulated cell, suggesting that the control of PRU proliferation could need other immune mediators such as TNFα, which interact with IFN-γ, as described in murine microglia [14]. Däubener et al. reported that interaction of IFN-γ and TNFα in human brain microvascular endothelial cells induces a strong induction of indoleamine 2,3-dioxygenase (IDO) [9]. Indoleamine 2,3-dioxygenase induction in Toxoplasma-infected human brain cells enhances tryptophane degradation and IFN-γ secretion which promote an anti-parasitic effect [8].
In conclusion, IFN-γ is implicated in the decrease of pro-inflammatory reactions in type II-infected microglial and endothelial cells, which promotes parasite persistance. In contrast, IFN-γ may have a role in controlling type I strain multiplication in human microglial and endothelial cells but not in human neurons. Contrary to what has been demonstrated in in vivo murine studies, our results show that the PRU strain proliferates in all stimulated and non-stimulated cells, as the result of a specific effect of this strain on host cell responses or of absence of the immune protein and cellular interaction that takes place in vivo. Further studies integrating these interactions would be needed to confirm these results.
Conflict of interest
None.
Acknowledgments
This work was supported by grants from the Conseil Régional du Limousin and “3IS, Infection et Immunité sous Immunosuppression” from FR 3503 Genomics Institute, Environment, Immunity, Health and Therapeutics (GEIST), Limoges, France. The authors would like to thank J. Cook-Moreau, H. Riahi, R. Mouzet and M. Gatet for their technical and scientific assistance.
References
- Barragan A, Brossier F, Sibley LD. 2005. Transepithelial migration of Toxoplasma gondii involves an interaction of intercellular adhesion molecule 1 (ICAM-1) with the parasite adhesin MIC2. Cellular Microbiology, 7(4), 561–568. [CrossRef] [PubMed] [Google Scholar]
- Brenier-Pinchart MP, Blanc-Gonnet E, Marche PN, Berger F, Durand F, Ambroise-Thomas P, Pelloux H. 2004. Infection of human astrocytes and glioblastoma cells with Toxoplasma gondii: monocyte chemotactic protein-1 secretion and chemokine expression in vitro. Acta Neuropathologica, 107(3), 245–249. [CrossRef] [PubMed] [Google Scholar]
- Brenier-Pinchart MP, Vigan I, Jouvin-Marche E, Marche PN, Pelet E, Gross U, Ambroise-Thomas P, Pelloux H. 2002. Monocyte chemotactic protein-1 secretion and expression after Toxoplasma gondii infection in vitro depend on the stage of the parasite. FEMS Microbiology Letters, 214(1), 45–49. [CrossRef] [PubMed] [Google Scholar]
- Cassaing S, Bessieres MH, Berry A, Berrebi A, Fabre R, Magnaval JF. 2006. Comparison between two amplification sets for molecular diagnosis of toxoplasmosis by real-time PCR. Journal of Clinical Microbiology, 44(3), 720–724. [CrossRef] [PubMed] [Google Scholar]
- Cekanaviciute E, Dietrich HK, Axtell RC, Williams AM, Egusquiza R, Wai KM, Koshy AA, Buckwalter MS. 2014. Astrocytic TGF-beta signaling limits inflammation and reduces neuronal damage during central nervous system Toxoplasma infection. Journal of Immunology, 193(1), 139–149. [CrossRef] [Google Scholar]
- Channon JY, Miselis KA, Minns LA, Dutta C, Kasper LH. 2002. Toxoplasma gondii induces granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor secretion by human fibroblasts: implications for neutrophil apoptosis. Infection and Immunity, 70(11), 6048–6057. [CrossRef] [PubMed] [Google Scholar]
- Combe CL, Curiel TJ, Moretto MM, Khan IA. 2005. NK cells help to induce CD8(+)-T-cell immunity against Toxoplasma gondii in the absence of CD4(+) T cells. Infection and Immunity, 73(8), 4913–4921. [CrossRef] [PubMed] [Google Scholar]
- Däubener W, Hadding U. 1997. Cellular immune reactions directed against Toxoplasma gondii with special emphasis on the central nervous system. Medical Microbiology Immunology, 185(4), 195–206. [CrossRef] [Google Scholar]
- Däubener W, Spors B, Hucke C, Adam R, Stins M, Kim KS, Schroten H. 2001. Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infection and Immunity, 69(10), 6527–6531. [CrossRef] [PubMed] [Google Scholar]
- Denkers EY, Gazzinelli RT, Martin D, Sher A. 1993. Emergence of NK1.1+ cells as effectors of IFN-gamma dependent immunity to Toxoplasma gondii in MHC class I-deficient mice. Journal of Experimental Medicine, 178(5), 1465–1472. [CrossRef] [Google Scholar]
- El-Shazly AM, Soliman M, El-Kalla MR, Rezk H, El-Nemr H, Handoussa AE, El-Aaty HE, Morsy TA. 2001. Evaluation of soluble adhesion molecules in the diagnosis of amoebiasis, giardiasis and toxoplasmosis. Journal of the Egyptian Society of Parasitology, 31(3), 691–700. [PubMed] [Google Scholar]
- Fischer HG, Nitzgen B, Reichmann G, Gross U, Hadding U. 1997. Host cells of Toxoplasma gondii encystation in infected primary culture from mouse brain. Parasitology Research, 83(7), 637–641. [CrossRef] [PubMed] [Google Scholar]
- Flores M, Saavedra R, Bautista R, Viedma R, Tenorio EP, Leng L, Sanchez Y, Juarez I, Satoskar AA, Shenoy AS, Terrazas LI, Bucala R, Barbi J, Satoskar AR, Rodriguez-Sosa M. 2008. Macrophage migration inhibitory factor (MIF) is critical for the host resistance against Toxoplasma gondii. FASEB Journal, 22(10), 3661–3671. [CrossRef] [Google Scholar]
- Freund YR, Zaveri NT, Javitz HS. 2001. In vitro investigation of host resistance to Toxoplasma gondii infection in microglia of BALB/c and CBA/Ca mice. Infection and Immunity, 69(2), 765–772. [CrossRef] [PubMed] [Google Scholar]
- Halonen SK, Chiu F, Weiss LM. 1998. Effect of cytokines on growth of Toxoplasma gondii in murine astrocytes. Infection and Immunity, 66(10), 4989–4993. [PubMed] [Google Scholar]
- Janabi N, Peudenier S, Heron B, Ng KH, Tardieu M. 1995. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neuroscience Letters, 195(2), 105–108. [CrossRef] [PubMed] [Google Scholar]
- Jebbari H, Roberts CW, Ferguson DJ, Bluethmann H, Alexander J. 1998. A protective role for IL-6 during early infection with Toxoplasma gondii. Parasite Immunology, 20(5), 231–239. [CrossRef] [PubMed] [Google Scholar]
- Luder CG, Giraldo-Velasquez M, Sendtner M, Gross U. 1999. Toxoplasma gondii in primary rat CNS cells: differential contribution of neurons, astrocytes, and microglial cells for the intracerebral development and stage differentiation. Experimental Parasitology, 93(1), 23–32. [CrossRef] [PubMed] [Google Scholar]
- Mammari N, Vignoles P, Halabi MA, Dardé ML, Courtioux B. 2014. In vitro infection of human nervous cells by two strains of Toxoplasma gondii: a kinetic analysis of immune mediators and parasite multiplication. PLoS One, 9(6), e98491. [CrossRef] [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Hu S, Chao CC. 1995. Human astrocytes inhibit intracellular multiplication of Toxoplasma gondii by a nitric oxide-mediated mechanism. Journal of Infectious Diseases, 171(2), 516–518. [CrossRef] [Google Scholar]
- Ross RA, Spengler BA, Biedler JL. 1983. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. Journal of the National Cancer Institute, 71(4), 741–747. [PubMed] [Google Scholar]
- Rozenfeld C, Martinez R, Seabra S, Sant’anna C, Goncalves JG, Bozza M, Moura-Neto V, De Souza W. 2005. Toxoplasma gondii prevents neuron degeneration by interferon-gamma-activated microglia in a mechanism involving inhibition of inducible nitric oxide synthase and transforming growth factor-beta1 production by infected microglia. American Journal of Pathology, 167(4), 1021–1031. [CrossRef] [Google Scholar]
- Schuindt SH, Oliveira BC, Pimentel PM, Resende TL, Retamal CA, DaMatta RA, Seipel D, Arnholdt AC. 2012. Secretion of multi-protein migratory complex induced by Toxoplasma gondii infection in macrophages involves the uPA/uPAR activation system. Veterinary Parasitology, 186(3–4), 207–215. [CrossRef] [PubMed] [Google Scholar]
- Schweitzer KM, Vicart P, Delouis C, Paulin D, Drager AM, Langenhuijsen MM, Weksler BB. 1997. Characterization of a newly established human bone marrow endothelial cell line: distinct adhesive properties for hematopoietic progenitors compared with human umbilical vein endothelial cells. Laboratory Investigation, 76(1), 25–36. [Google Scholar]
- Suzuki Y, Claflin J, Wang X, Lengi A, Kikuchi T. 2005. Microglia and macrophages as innate producers of interferon-gamma in the brain following infection with Toxoplasma gondii. International Journal of Parasitology, 35(1), 83–90. [CrossRef] [Google Scholar]
- Suzuki Y, Orellana MA, Schreiber RD, Remington JS. 1988. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science, 240(4851), 516–518. [CrossRef] [PubMed] [Google Scholar]
Cite this article as: Mammari N, Vignoles P, Halabi MA, Dardeé M-L & Courtioux B: Interferon gamma effect on immune mediator production in human nerve cells infected by two strains of Toxoplasma gondii. Parasite, 2015, 22, 39.
All Tables
Cell types, parasite strains and IFN-γ effects on pro-inflammatory protein expressions. Pro-inflammatory proteins were analysed in supernatants of cultivated RH- or PRU-infected cells using Proteome Profiler Array. All spots were quantified as pixel units by GeneTools. Quantification corresponds to the pixel values of uninfected cells (negative control) subtracted from pixel values of infected cells. The effect level was determined using the Scheirer-Ray-Hare test and Steel-Dwass and/or Siegel-Castellan post hoc tests. ↘ expression decrease with p < 0.05; ↘↘ expression decrease with p < 0.001. ↗ expression increase with p < 0.05; ↗↗ expression increase with p < 0.001.
All Figures
|
Figure 1. Effect of IFN-γ on parasite multiplication. Microglial cells, endothelial cells and neurons were stimulated by IFN-γ before being infected with the RH or PRU strain of T. gondii for 24 h. Parasite burden was quantified by semi-quantitative RT-PCR in stimulated and infected cells (black) and in non-stimulated and infected cells (grey). Parasite quantification was evaluated as follows: final number of tachyzoites/initial number of tachyzoites. Comparison was performed using the Scheirer-Ray-Hare test. **p < 0.01. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.